

Transcriptome Sequencing and Analysis of Trichoderma polysporum Infection in Avena fatua L. Leaves before and after Infection

Abstract
1. Introduction
2. Materials and Methods
2.1. Test Strains
2.2. Infection of A. fatua L. with T. polysporum HZ-31 and Culture Treatments
2.3. RNA Extraction and Quality Testing of Samples
2.4. Library Construction and Sequencing
2.5. Analysis of Transcriptome Sequencing Information
2.5.1. Data Quality Control
2.5.2. Reference Sequence Comparison Analysis
2.5.3. Expression Statistics
2.5.4. Differential Expression Analysis
2.5.5. Differential Gene Enrichment Analysis and Functional Annotation
2.5.6. Validation of Transcriptome Results
3. Results and Analysis
3.1. Transcriptome Data Assembly Results
3.2. Reference T. polysporum Genome Comparison Results
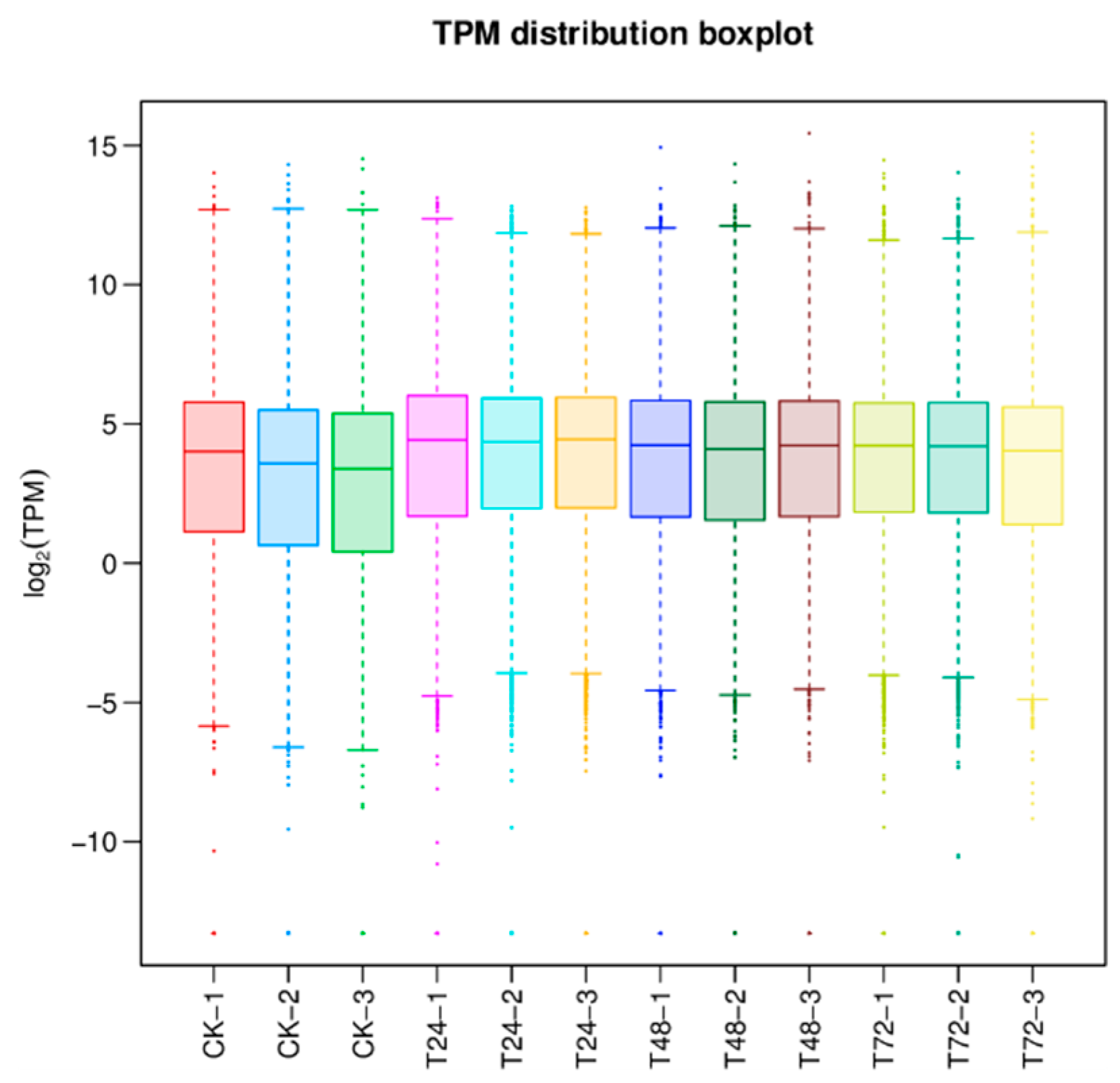
3.3. Expression Analysis
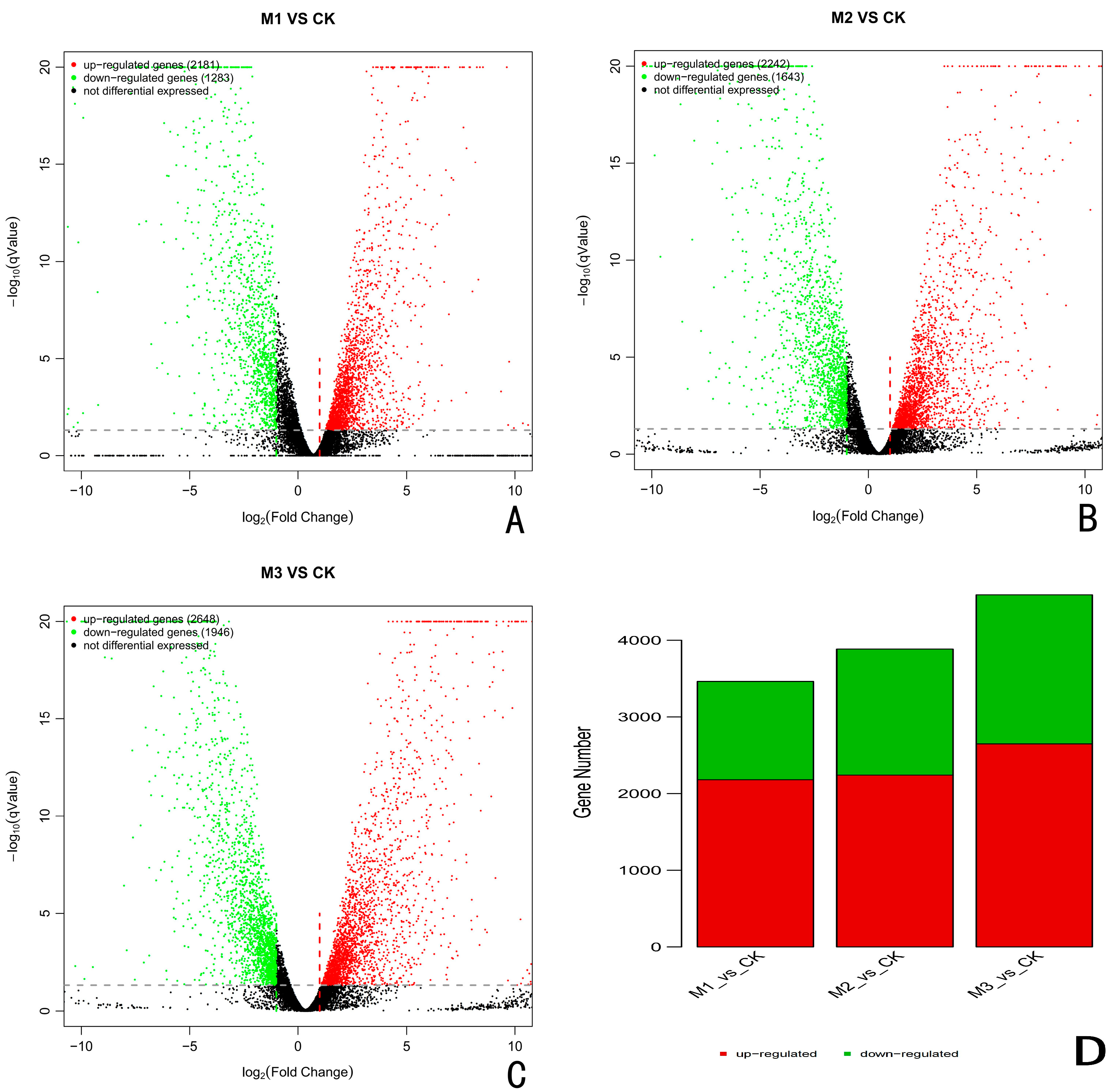
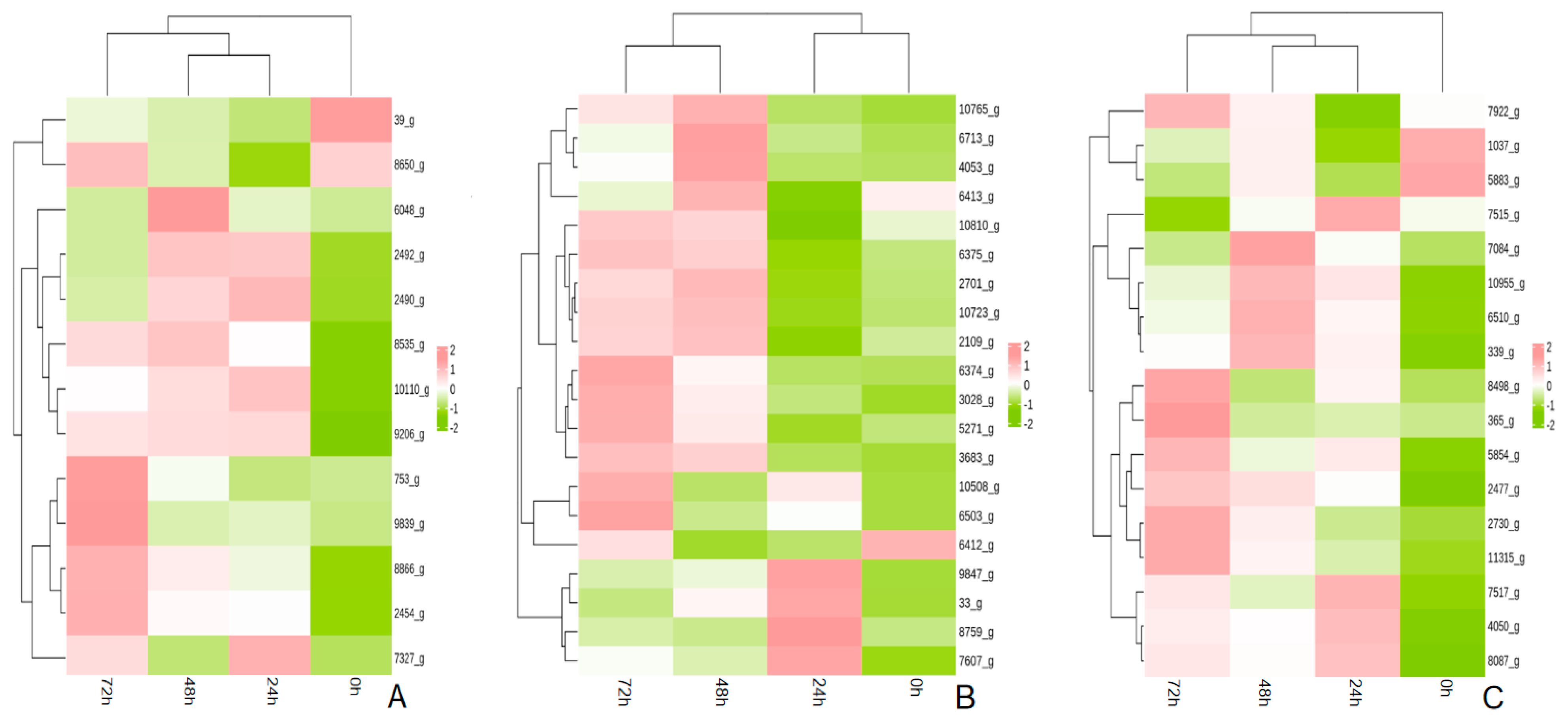
3.4. Differential Gene Analysis
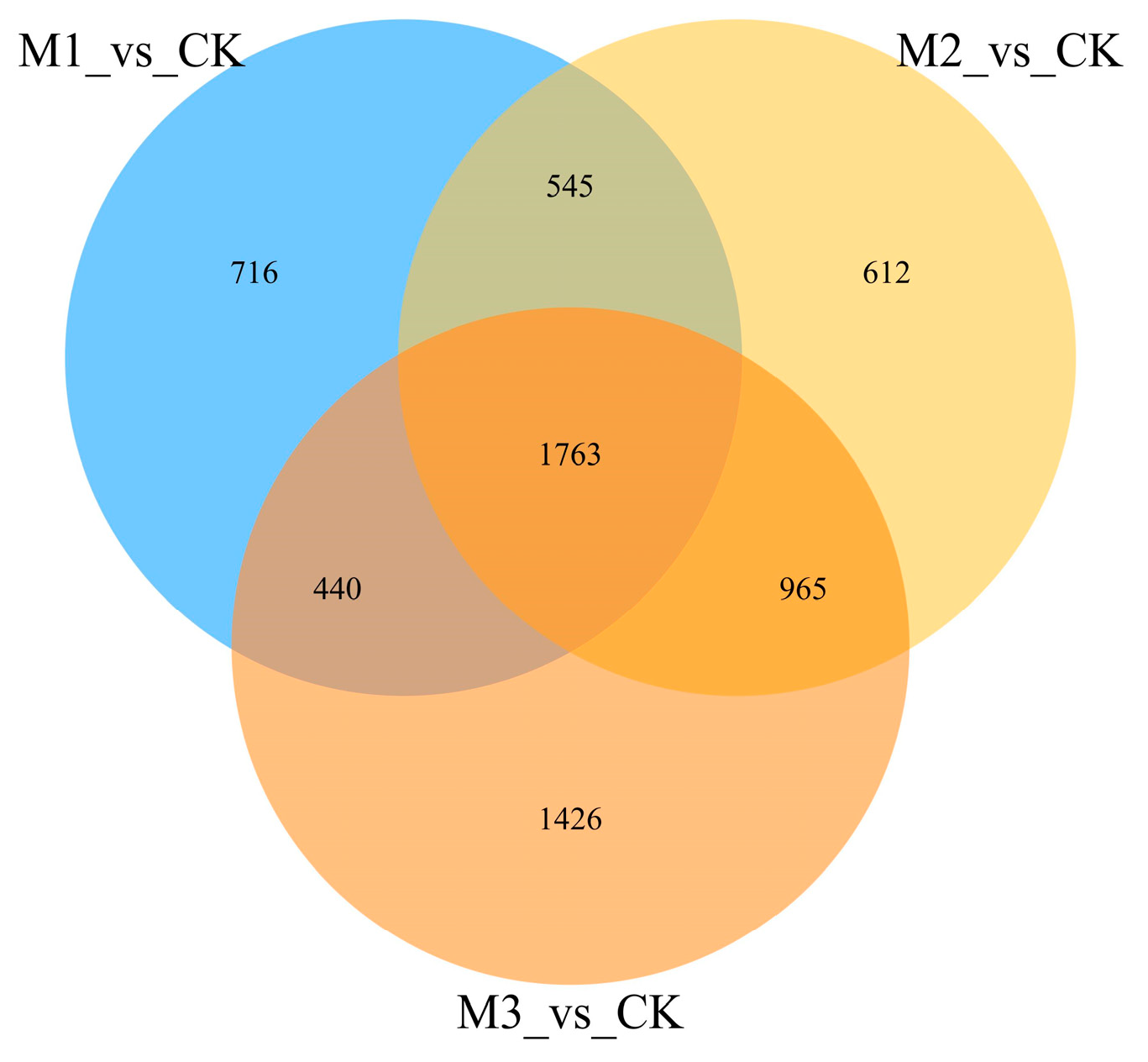
3.5. Venn Analysis
3.6. Enrichment Analysis of Up-Regulated Differentially Expressed Genes
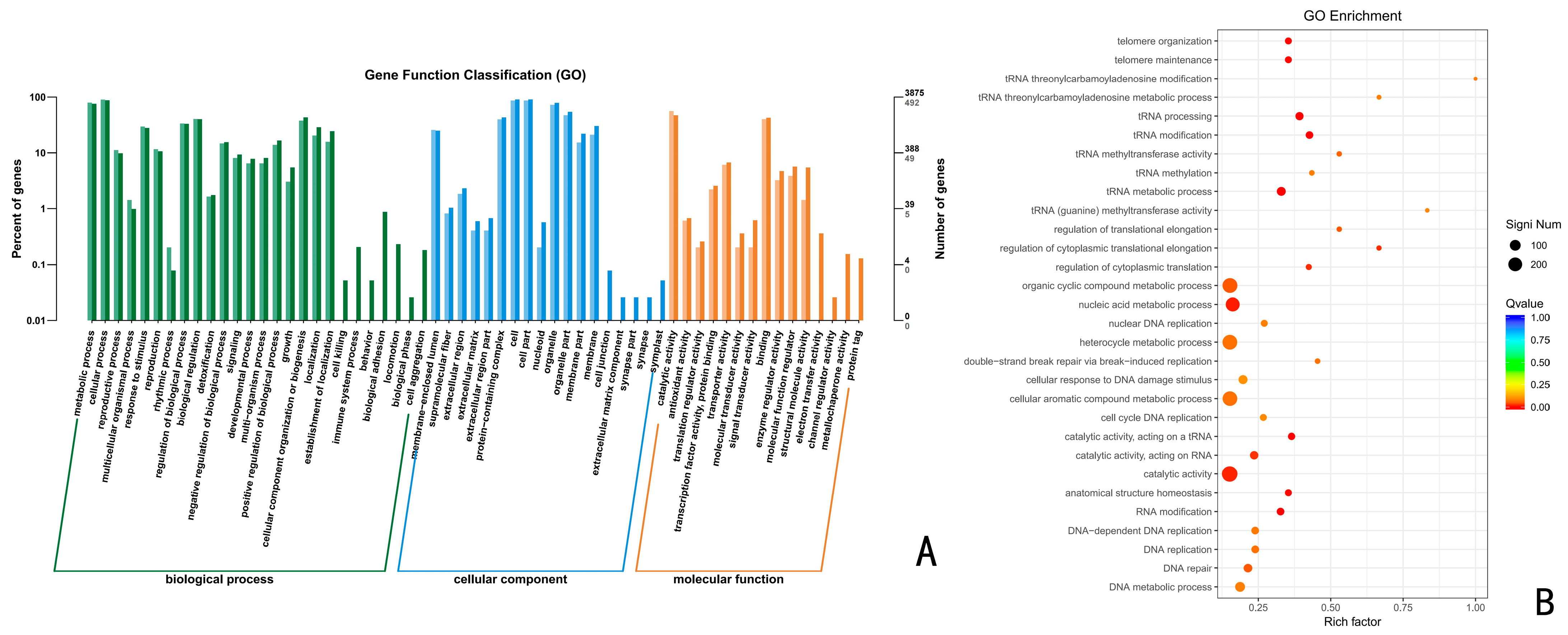
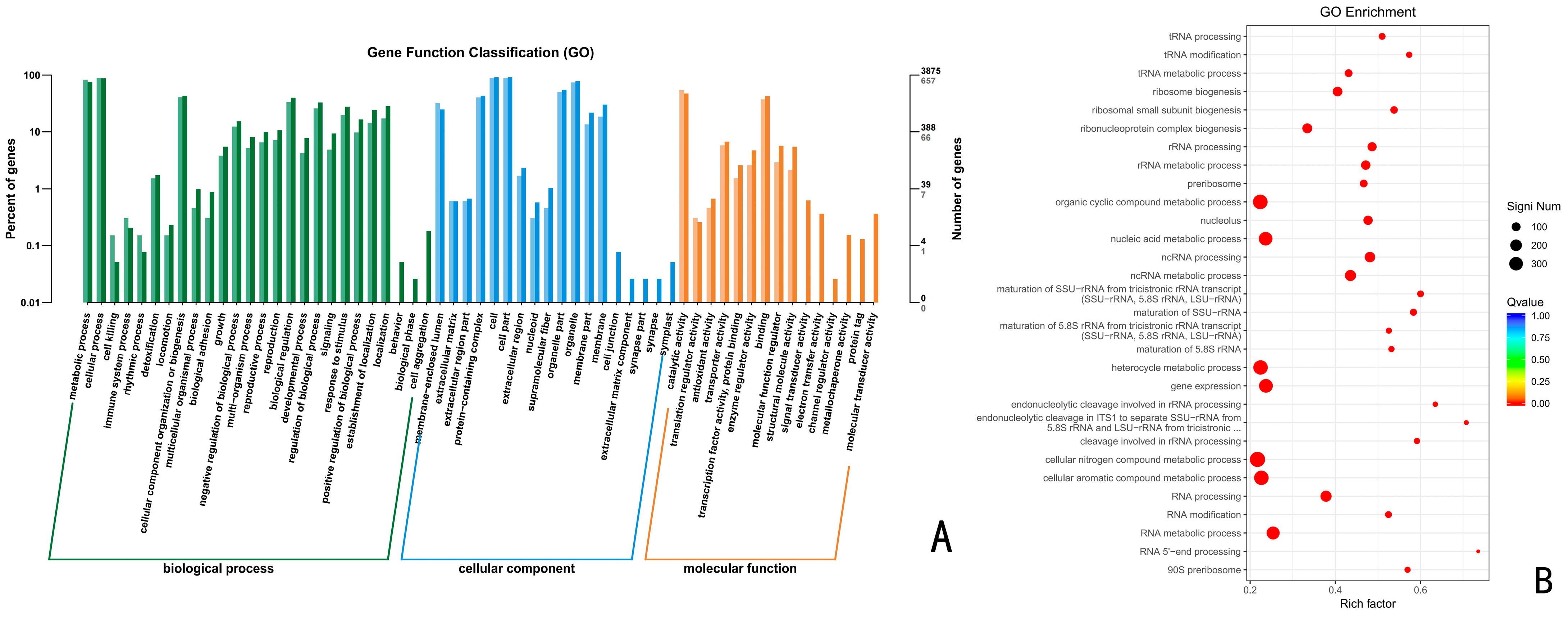
3.6.1. GO Functional Enrichment Analysis of Up-Regulated Differentially Expressed Genes
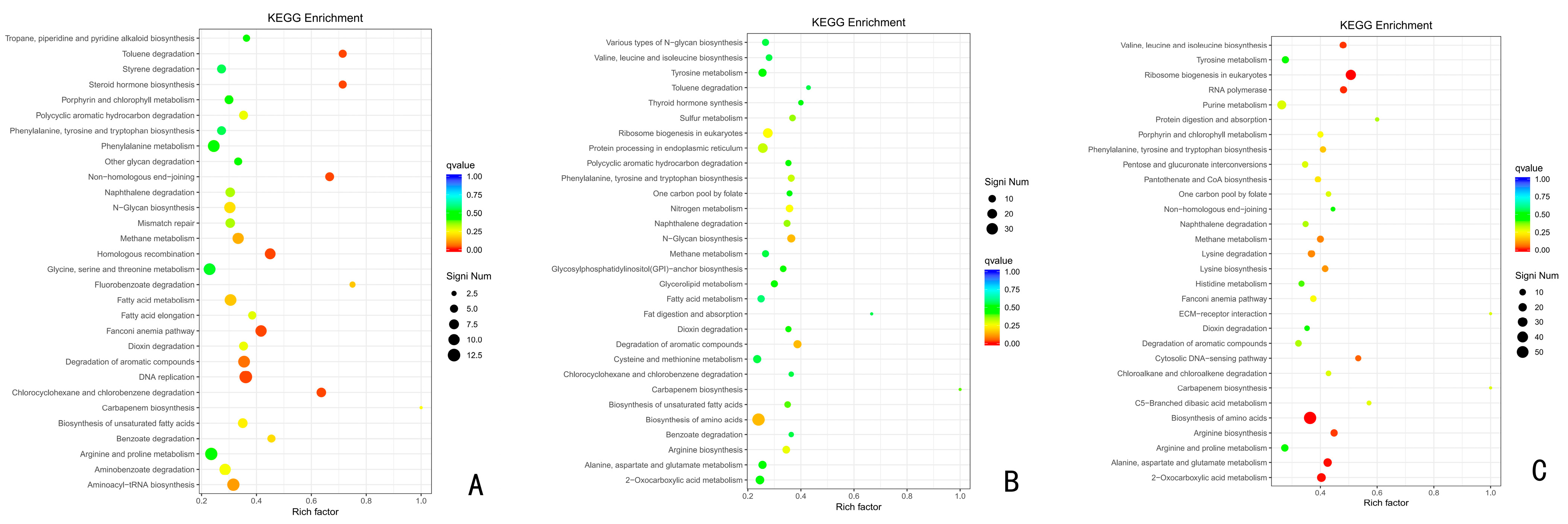
3.6.2. KEGG Functional Enrichment Analysis of Up-Regulated Differentially Expressed Genes
3.6.3. Metabolic Pathway Analysis
3.7. Validation of Transcriptome Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Iannucci, A.; Fragasso, M.; Platani, C.; Papa, R. Plant growth and phenolic compounds in the rhizosphere soil of wild oat (Avena fatua L.). Front. Plant Sci. 2013, 4, 509. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, A.; Sharma, R.; Sindhu, S.; Sindhu, S.S. Resource partitioning in the rhizosphere by inoculated Bacillus spp. towards growth stimulation of wheat and suppression of wild oat (Avena fatua L.) weed. Physiol. Mol. Biol. Plants 2019, 25, 1483–1495. [Google Scholar] [CrossRef]
- Chauhan, B.S. The world’s first glyphosate-resistant case of Avena fatua L. and Avena sterilis ssp. ludoviciana (Durieu) Gillet & Magne and alternative herbicide options for their control. PLoS ONE 2022, 17, e0262494. [Google Scholar]
- Li, W.; Shen, S.; Chen, H. Bio-herbicidal potential of wheat rhizosphere bacteria on Avena fatua L. grass. Bioengineered 2021, 1, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Tafoya-Razo, J.A.; Mora-Munguía, S.A.; Torres-García, J.R. Diversity of Herbicide-Resistance Mechanisms of Avena fatua L. to Acetyl-CoA Carboxylase-Inhibiting Herbicides in the Bajio, Mexico. Plants 2022, 11, 1644. [Google Scholar] [CrossRef] [PubMed]
- Kamo, M.; Tojo, M.; Yamazaki, Y.; Itabashi, T.; Takeda, H.; Wakana, D.; Hosoe, T. Isolation of growth inhibitors of the snow rot pathogen Pythium iwayamai from an arctic strain of Trichoderma polysporum. J. Antibiot. 2016, 69, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chaturvedi, V.; Chaturvedi, S. Novel Trichoderma polysporum Strain for the Biocontrol of Pseudogymnoascus destructans, the Fungal Etiologic Agent of Bat White Nose Syndrome. PLoS ONE 2015, 10, e0141316. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Lasek-Nesselquist, E.; Chaturvedi, V.; Chaturvedi, S. Trichoderma polysporum selectively inhibits white-nose syndrome fungal pathogen Pseudogymnoascus destructans amidst soil microbes. Microbiome 2018, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, Y.; Mishra, R.C.; Bae, H. Fungal endophytes inhabiting mountain-cultivated ginseng (Panax ginseng Meyer): Diversity and biocontrol activity against ginseng pathogens. Sci. Rep. 2017, 7, 16221. [Google Scholar] [CrossRef]
- Eziashi, E.; Uma, N.U.; Adekunle, A.A.; Airede, C.E.; Odigie, E.E. Evaluation of lyophilized and non lyophilized toxins from Trichoderma species for the control of Ceratocystis paradoxa. Afr. J. Agric. Res. 2010, 5, 1733–1738. [Google Scholar]
- Zhu, H.X.; Ma, Y.Q.; Guo, Q.Y.; Xu, B.L. Biological weed control using Trichoderma polysporum strain HZ-31. Crop Prot. 2020, 134, 105161. [Google Scholar] [CrossRef]
- Rüegger, A.; Kuhn, M.; Lichti, H.; Loosli, H.R.; Huguenin, R.; Quiquerez, C.; von Wartburg, A. Cyclosporin A, ein immunsuppressiv wirksamer Peptidmetabolit aus Trichoderma polysporum (Link ex Pers.) Rifai [Cyclosporin A, a Peptide Metabolite from Trichoderma polysporum (Link ex Pers.) Rifai, with a remarkable immunosuppressive activity. Helv. Chim. Acta. 1976, 59, 1075–1092. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Iida, A.; Uesato, S.; Takaishi, Y.; Shingu, T.; Saito, M.; Morita, M. Structural elucidation of trichosporin-B-Ia, IIIa, IIId and V from Trichoderma polysporum. J. Antibiot. 1988, 41, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhao, P.; Zhao, L.; Jia, A.; Liu, C.; Zhang, L.; Xia, X. Anthraquinone Derivatives from a Sea Cucumber-Derived Trichoderma sp. Fungus with Antibacterial Activities. Chem. Nat. Compd. 2020, 56, 112–114. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, H.Y.; Ma, Y.Q.; Guo, Q.Y. Identification and extraction of herbicidal compounds from metabolites of Trichoderma polysporum HZ-31. Weed Sci. 2023, 71, 39–49. [Google Scholar] [CrossRef]
- Bhadauria, V.; Popescu, L.; Zhao, W.S.; Peng, Y.L. Fungal transcriptomics. Microbiol. Res. 2007, 162, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Xue, M.; Shen, Z.; Jia, X.; Hou, X.; Lai, D.; Zhou, L. Phytotoxic Secondary Metabolites from Fungi. Toxins 2021, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.; Zhu, X.; Wang, W. Genome and transcriptome sequencing of Trichoderma harzianum T4, an important biocontrol fungus of Rhizoctonia solani, reveals genes related to mycoparasitism. Can. J. Microbiol. 2024, 70, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.C.; Tang, W.; Ma, J.K.; Chen, J.W.; Yang, D.J.; Gao, F.Y.; Sun, H.J.; Xie, Y.P.; Zhang, C.J. Transcriptome analysis of Fusarium solani infected sweet potato. Acta Agric. Zhejiangensis 2023, 35, 1097–1107. [Google Scholar]
- Santiago, K.A.A.; Wong, W.C.; Goh, Y.K.; Tey, S.H.; Ting, A.S.Y. Pathogenicity of monokaryotic and dikaryotic mycelia of Ganoderma boninense revealed via LC-MS-based metabolomics. Sci. Rep. 2024, 14, 5330. [Google Scholar] [CrossRef]
- Zhu, M. Cloning and Functional Verification of Key Genes for L-Phenylalanine Biosynthesis in Rose. Master’s Thesis, Yangzhou University, Yangzhou, China, 2020. [Google Scholar]
- Wang, B. Metabolic Engineering of Engineered Strains of Phenol Biosynthesis. Master’s Thesis, Qufu Normal University, Qufu, China, 2016. [Google Scholar]
- Jin, J.Y.; Diao, Y.F.; Yu, C.M.; Xiong, X.; Zhao, T.; He, B.L.; Liu, H.X. Transcriptome analysis of the process of infectation of apple branches by Fusarium fusarium. J. Shandong Agric. Univ. (Nat. Sci. Ed.) 2021, 52, 187–193. [Google Scholar]
- Reid, M.F.; Fewson, C.A. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 1994, 20, 13–56. [Google Scholar] [CrossRef] [PubMed]
- de Smidt, O.; du Preez, J.C.; Albertyn, J. The alcohol dehydrogenases of Saccharomyces cerevisiae: A comprehensive review. FEMS Yeast Res. 2008, 8, 967–978. [Google Scholar] [CrossRef] [PubMed]
- DafaAlla, T.E.I.M.; Abdalla, M.; El-Arabey, A.A.; Eltayb, W.A.; Mohapatra, R.K. Botrytis cinerea alcohol dehydrogenase mediates fungal development, environmental adaptation and pathogenicity. J. Biomol. Struct. Dyn. 2022, 40, 12426–12438. [Google Scholar] [CrossRef] [PubMed]
- Prosperini, A.; Berrada, H.; Ruiz, M.J.; Caloni, F.; Coccini, T.; Spicer, L.J.; Perego, M.C.; Lafranconi, A. A Review of the Mycotoxin Enniatin, B. Front. Public Health 2017, 5, 304. [Google Scholar] [CrossRef]
- Herrmann, M.; Zocher, R.; Haese, A. Enniatin production by fusarium strains and its effect on potato tuber tissue. Appl. Environ. Microbiol. 1996, 62, 393–398. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Size (bp) |
|---|---|---|---|
| actin | CCTCCTCTTCCTTGCCAGCATTG | CCTCCTCTTCCTTGCCAGCATTG | 111 |
| Unigene2109 | TGGCGGTGGTATTGGAGGTCTG | TCCTTGTATGCTGGTGCTTGTTCG | 102 |
| Unigene6375 | ATTCTGAACGACCGTGAACTCTTGG | TGGTGTGGAAGCATCGCATGTG | 96 |
| Unigene8535 | GCTGACTGGCAATGCTCTATGAC | CAATGGGCTGGGCGGTCTC | 139 |
| Unigene5854 | CAATGGGCTGGGCGGTCTC | CAGCGTTCGGAGTGTTGATGG | 139 |
| Unigene3028 | TCTCGCTCGCTTGATGGAAGG | ATGAGGAGTTGGAGGTCGTATCG | 119 |
| Unigene4053 | CTTGGCTTAGAATCCCTCGTTGAC | TGATGGTGCCGCCGTTCTC | 136 |
| Unigene5271 | GATGGCTTCTCCTCTGCTATGTG | TCTGAACTCCAATATGTCCCAAACC | 134 |
| Unigene6412 | CGAAGATGCTGCTGCGTTAGG | CGAACAGTATCATACACGGCTAGG | 90 |
| Unigene6713 | TGTTGCGAATGAGCGAGATGC | CCCTTGTAGTTTGTTCCTCCGTTAG | 99 |
| Unigene339 | GGTTCTCTCTGGCGGTTTCTTG | CCTCGTTAGCATGTGCGTAGTC | 80 |
| Unigene2759 | TGGCGGACTTGGTGGATGAG | TGCGTGATATTGATAGAGGCTTGC | 84 |
| Unigene3429 | TGCGAACTGGACTGGACTCTG | GTTGATCTCGGTGACGGCTTTC | 110 |
| Unigene5483 | TCGCCTATGCCGTGGTTCC | GGTTCGTCCATTCGCCAGATG | 150 |
| Unigene7084 | GGACCATGAGACCTGATAGCATTG | AGTCGCTGGCTCGGTTACG | 137 |
| Unigene259 | CAATGCCGCCGATTCACAGG | GGACCTTGTAATAGTTGCCGTATGG | 92 |
| Unigene8087 | AGGTGTTGGCGAGGAGTATCAG | TCAAGCGTCTCTTCAGTCTTTAGTG | 81 |
| Unigene2730 | GCCATTGAATGCCTTGCTTGAC | ATGTCCGCCGCCTTGGTAG | 102 |
| Unigene753 | TCGCTCGGTGTCGCCATC | AAGCAGTCTTCGTTACCTGTTGTG | 116 |
| Unigene6048 | TCGCCTCGGAGCAGATTGTC | GTTCATCAGCCATCGCAGGTAG | 143 |
| Unigene10110 | GGCGGCGGCATTGTATTCG | GCGGATTGCTGCTGGTCATAG | 125 |
| Sample | Raw Reads | Clean Reads | Clean Bases | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| CK-1 | 64,467,532 | 62,859,772 | 9,198,358,055 | 99.00% | 96.17% | 52.39% |
| CK-2 | 61,689,040 | 60,084,542 | 8,787,122,185 | 98.98% | 96.16% | 52.54% |
| CK-3 | 55,680,734 | 54,362,330 | 7,879,606,421 | 99.07% | 96.47% | 53.05% |
| T24-1 | 56,045,126 | 54,310,362 | 7,791,837,914 | 98.85% | 95.69% | 52.06% |
| T24-2 | 55,269,126 | 54,006,616 | 7,771,363,231 | 99.03% | 96.29% | 52.61% |
| T24-3 | 61,983,022 | 60,503,534 | 8,682,351,056 | 99.04% | 96.32% | 52.66% |
| T48-1 | 57,216,160 | 55,900,958 | 8,163,997,746 | 99.03% | 96.31% | 51.87% |
| T48-2 | 49,386,632 | 48,061,810 | 7,007,420,562 | 98.97% | 96.09% | 51.68% |
| T48-3 | 51,854,456 | 50,528,528 | 7,354,822,322 | 99.01% | 96.19% | 51.68% |
| T72-1 | 77,526,690 | 75,760,238 | 10,964,415,778 | 99.06% | 96.41% | 52.34% |
| T72-2 | 59,115,428 | 57,610,338 | 8,362,733,753 | 99.01% | 96.24% | 52.51% |
| T72-3 | 58,408,616 | 56,724,148 | 8,171,046,956 | 98.93% | 95.97% | 51.80% |
| Sample | Clean Reads | Total Mapped | Multiple Mapped | Uniquely Mapped | Read1 Mapped | Read2 Mapped | Proper Mapped |
|---|---|---|---|---|---|---|---|
| CK-1 | 56,568,506 (100.00%) | 56,110,923 (99.19%) | 163,743 (0.29%) | 55,947,180 (98.90%) | 27,979,165 (49.46%) | 27,968,015 (49.44%) | 54,648,680 (96.61%) |
| CK-2 | 59,334,990 (100.00%) | 58,694,852 (98.92%) | 238,005 (0.40%) | 58,456,847 (98.52%) | 29,220,950 (49.25%) | 29,235,897 (49.27%) | 57,088,794 (96.21%) |
| CK-3 | 54,102,082 (100.00%) | 53,611,134 (99.09%) | 176,271 (0.33%) | 53,434,863 (98.77%) | 26,719,566 (49.39%) | 26,715,297 (49.38%) | 51,866,660 (95.87%) |
| T24-1 | 50,455,296 (100.00%) | 49,908,731 (98.92%) | 147,119 (0.29%) | 49,761,612 (98.63%) | 24,899,621 (49.35%) | 24,861,991 (49.28%) | 48476952 (96.08%) |
| T24-2 | 53,627,174 (100.00%) | 52,963,908 (98.76%) | 179,316 (0.33%) | 52,784,592 (98.43%) | 26,400,523 (49.23%) | 26,384,069 (49.20%) | 50,871,496 (94.86%) |
| T24-3 | 59,771,456 (100.00%) | 59,042,883 (98.78%) | 209,930 (0.35%) | 58,832,953 (98.43%) | 29,428,530 (49.24%) | 29,404,423 (49.19%) | 56,860,444 (95.13%) |
| T48-1 | 52,454,282 (100.00%) | 51,511,562 (98.20%) | 238,042 (0.45%) | 51,273,520 (97.75%) | 25,640,147 (48.88%) | 25,633,373 (48.87%) | 49,681,412 (94.71%) |
| T48-2 | 34,189,154 (100.00%) | 33,535,178 (98.09%) | 114,753 (0.34%) | 33,420,425 (97.75%) | 16,714,982 (48.89%) | 16,705,443 (48.86%) | 32,285,094 (94.43%) |
| T48-3 | 29,116,484 (100.00%) | 27,919,427 (95.89%) | 94,113 (0.32%) | 27,825,314 (95.57%) | 13,918,967 (47.80%) | 13,906,347 (47.76%) | 26,793,860 (92.02%) |
| T72-1 | 75,273,746 (100.00%) | 74,106,054 (98.45%) | 345,027 (0.46%) | 73,761,027 (97.99%) | 36,888,884 (49.01%) | 36,872,143 (48.98%) | 71,239,844 (94.64%) |
| T72-2 | 57,056,046 (100.00%) | 56,079,845 (98.29%) | 281,043 (0.49%) | 55,798,802 (97.80%) | 27,908,963 (48.91%) | 27,889,839 (48.88%) | 53,954,714 (94.56%) |
| T72-3 | 54,409,280 (100.00%) | 53,175,243 (97.73%) | 320,973 (0.59%) | 52,854,270 (97.14%) | 26,441,123 (48.60%) | 26,413,147 (48.55%) | 51,321,778 (94.33%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; He, Y. Transcriptome Sequencing and Analysis of Trichoderma polysporum Infection in Avena fatua L. Leaves before and after Infection. J. Fungi 2024, 10, 346. https://doi.org/10.3390/jof10050346
Zhu H, He Y. Transcriptome Sequencing and Analysis of Trichoderma polysporum Infection in Avena fatua L. Leaves before and after Infection. Journal of Fungi. 2024; 10(5):346. https://doi.org/10.3390/jof10050346
Chicago/Turabian StyleZhu, Haixia, and Yushan He. 2024. "Transcriptome Sequencing and Analysis of Trichoderma polysporum Infection in Avena fatua L. Leaves before and after Infection" Journal of Fungi 10, no. 5: 346. https://doi.org/10.3390/jof10050346
APA StyleZhu, H., & He, Y. (2024). Transcriptome Sequencing and Analysis of Trichoderma polysporum Infection in Avena fatua L. Leaves before and after Infection. Journal of Fungi, 10(5), 346. https://doi.org/10.3390/jof10050346