
The Known and Unknown “Knowns” of Human Susceptibility to Coccidioidomycosis

Abstract
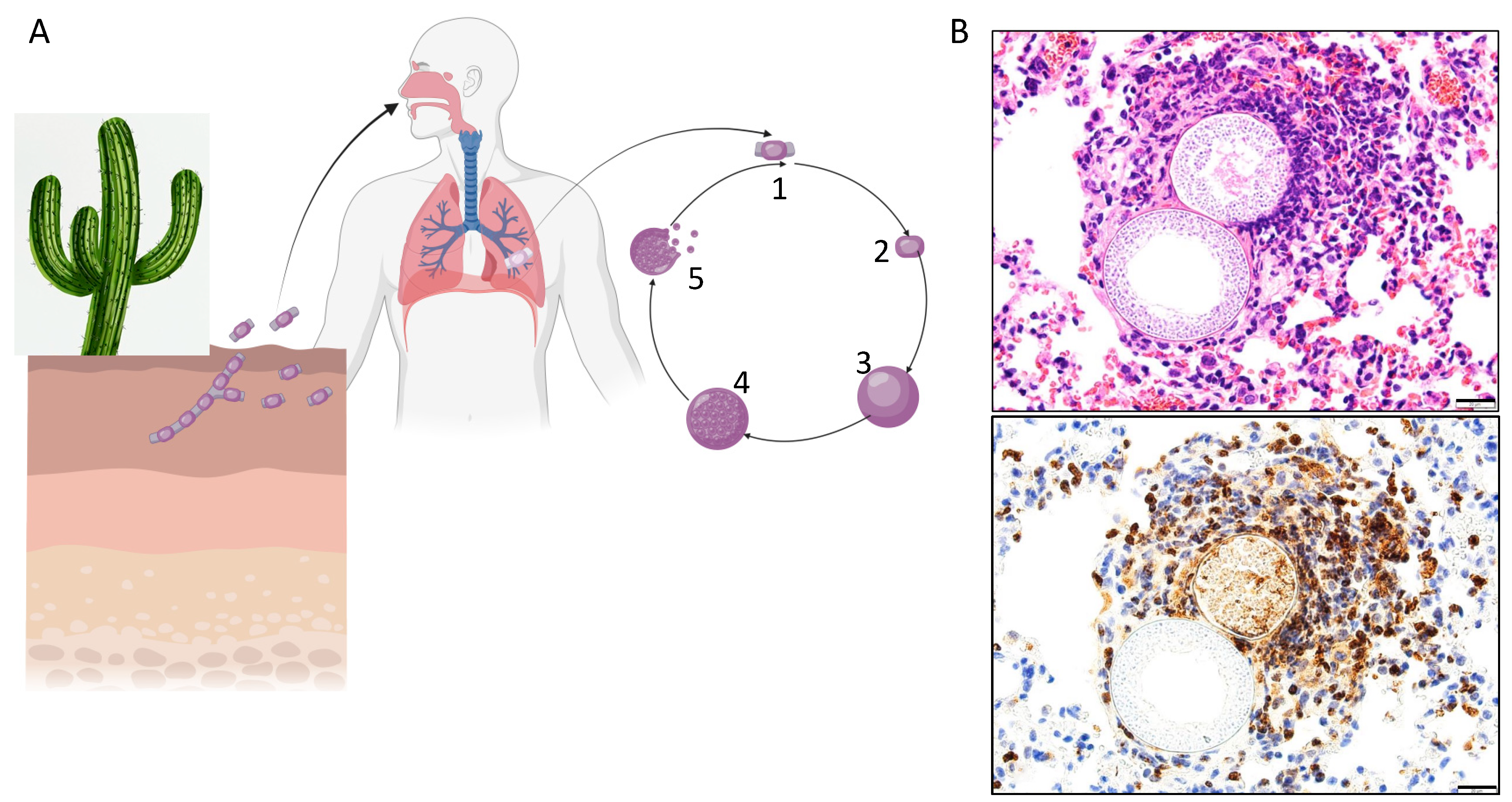
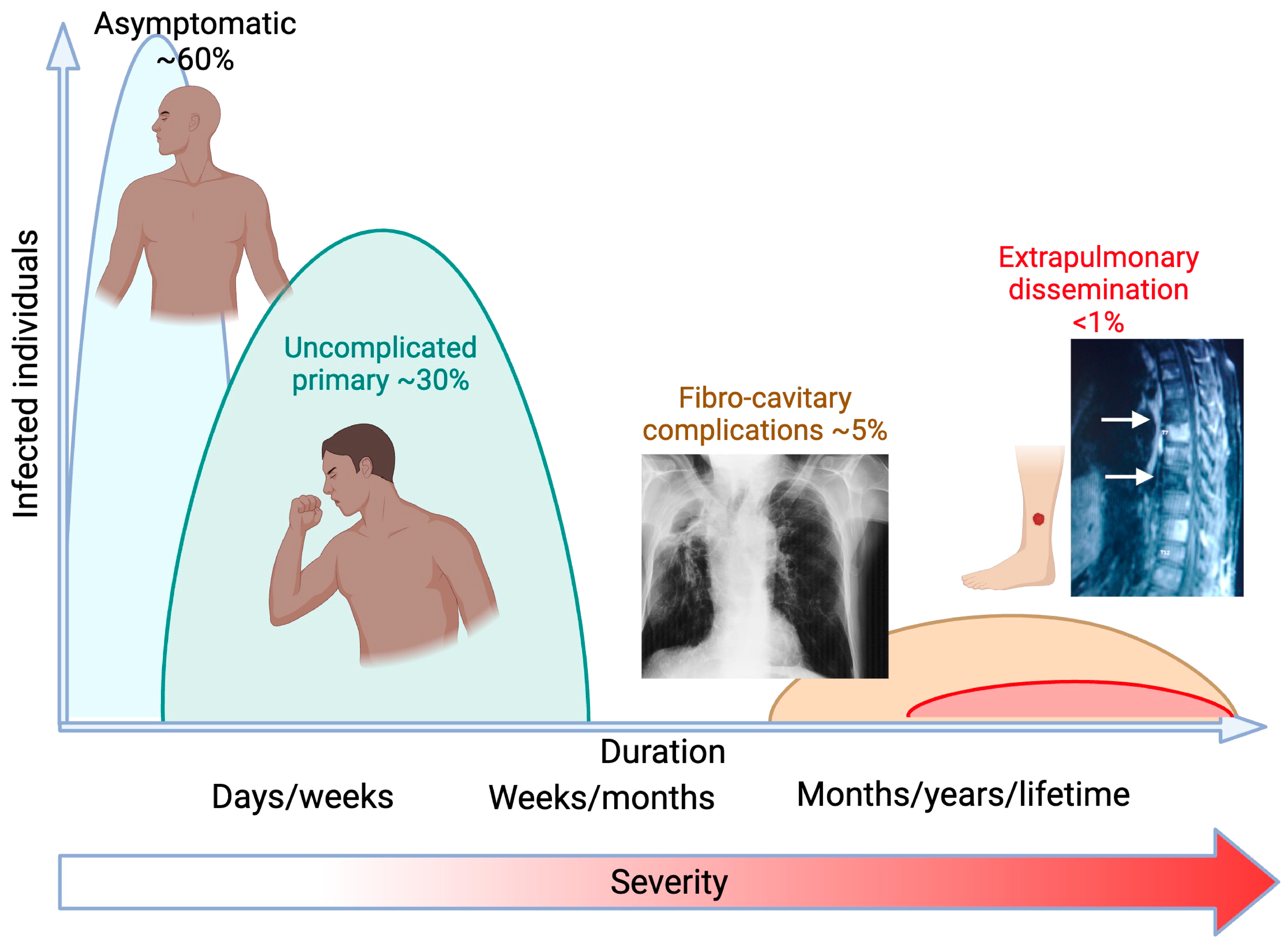
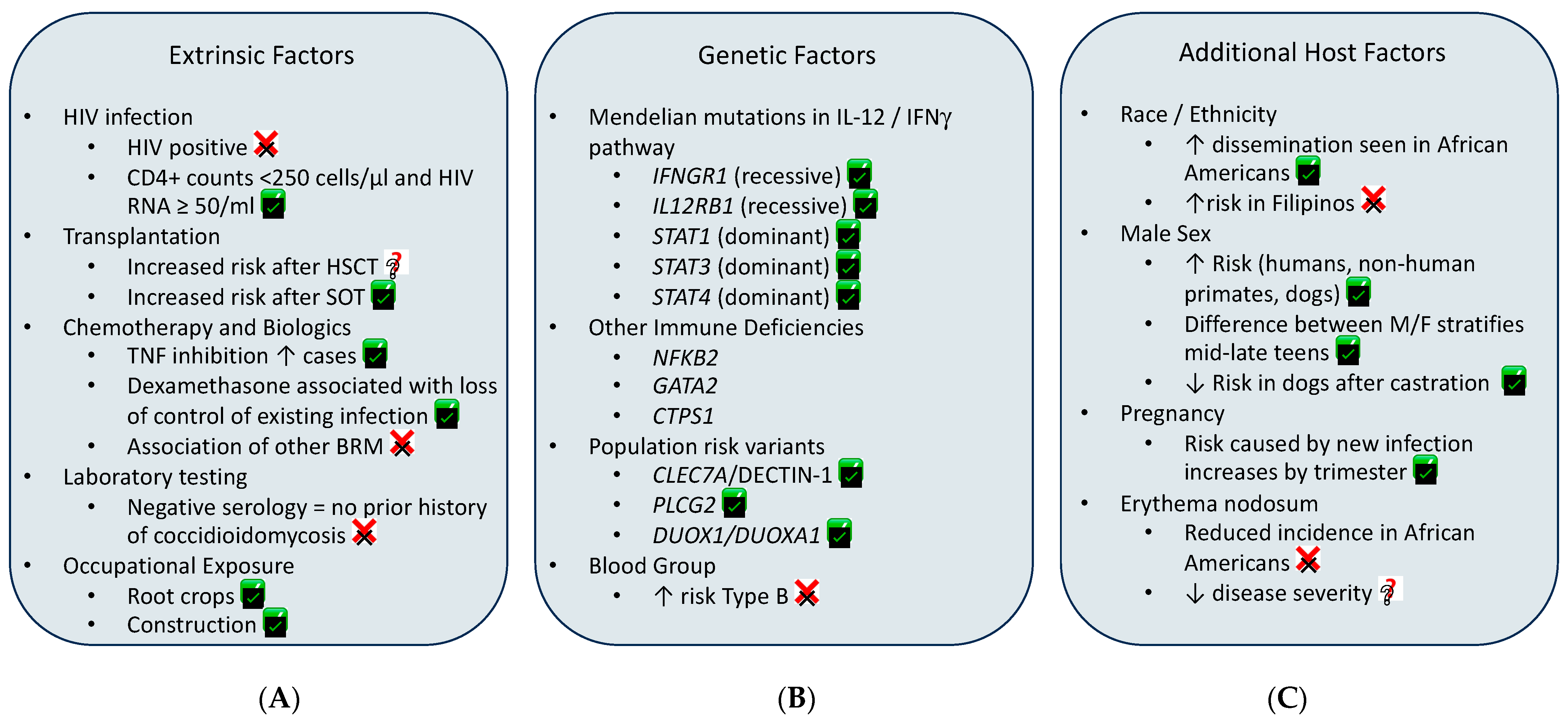
1. Background
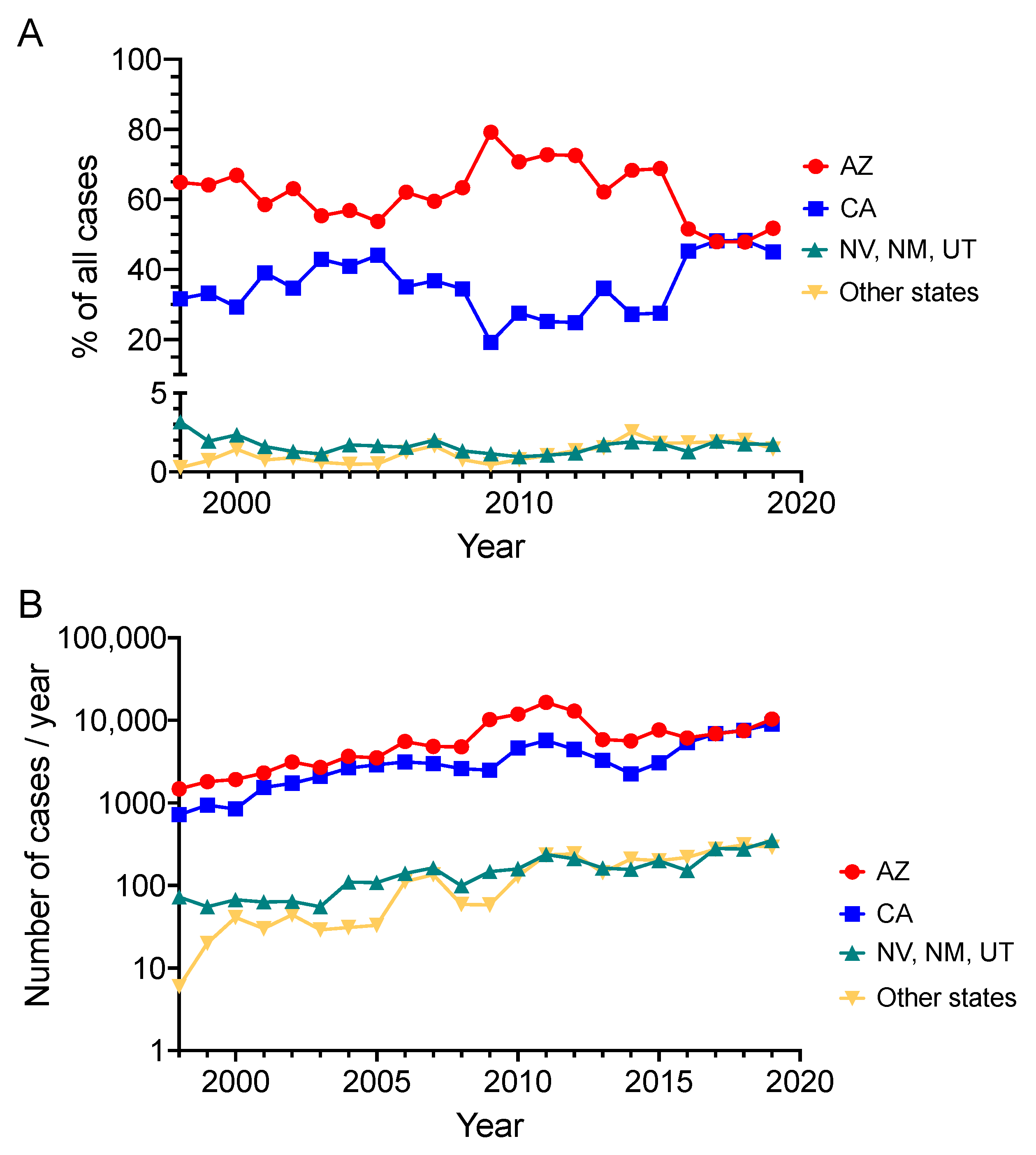
2. Exogenous Risks of Infection and Disseminated Disease
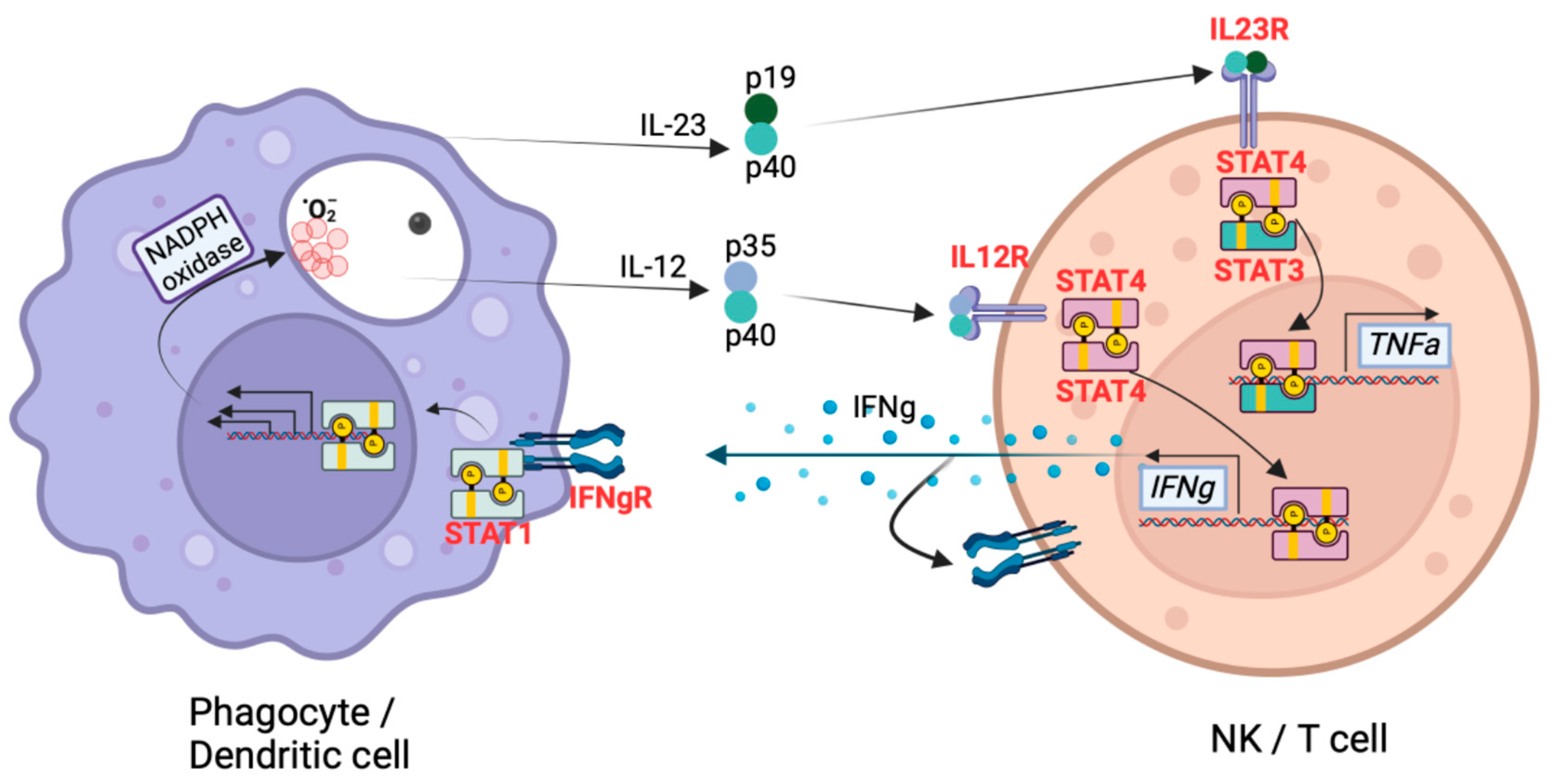
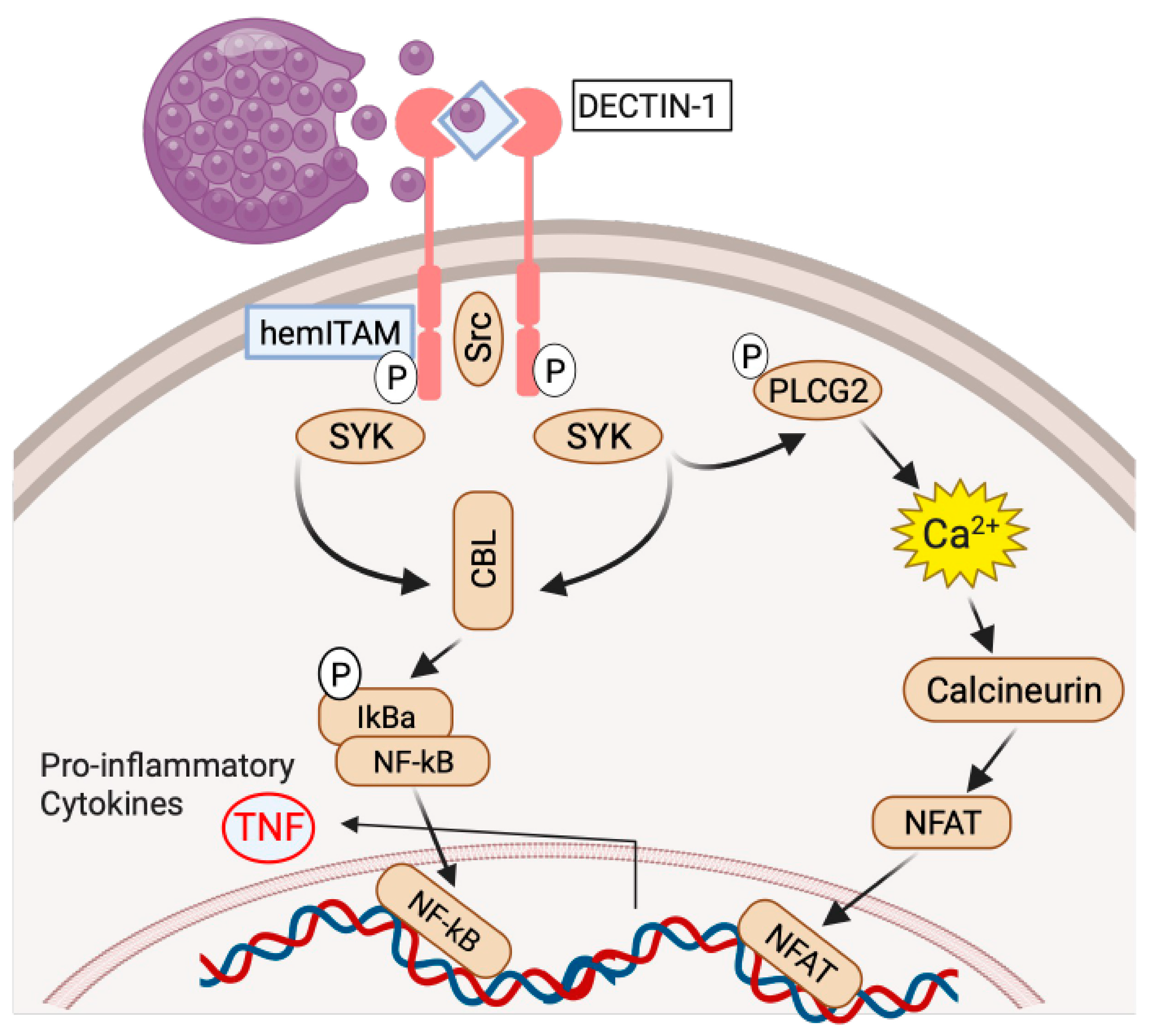
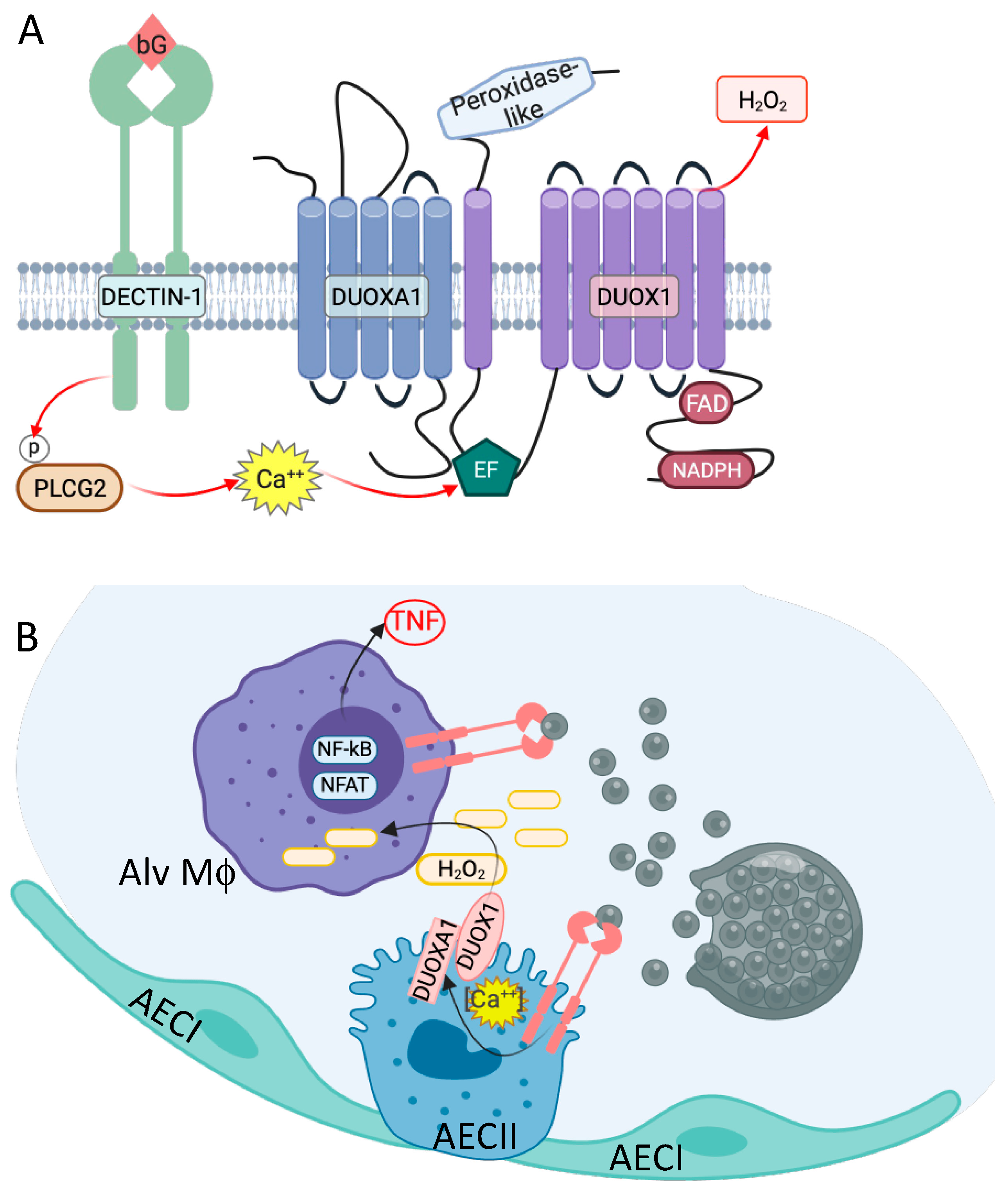
3. Genetic Variants Associated with Coccidioides Dissemination
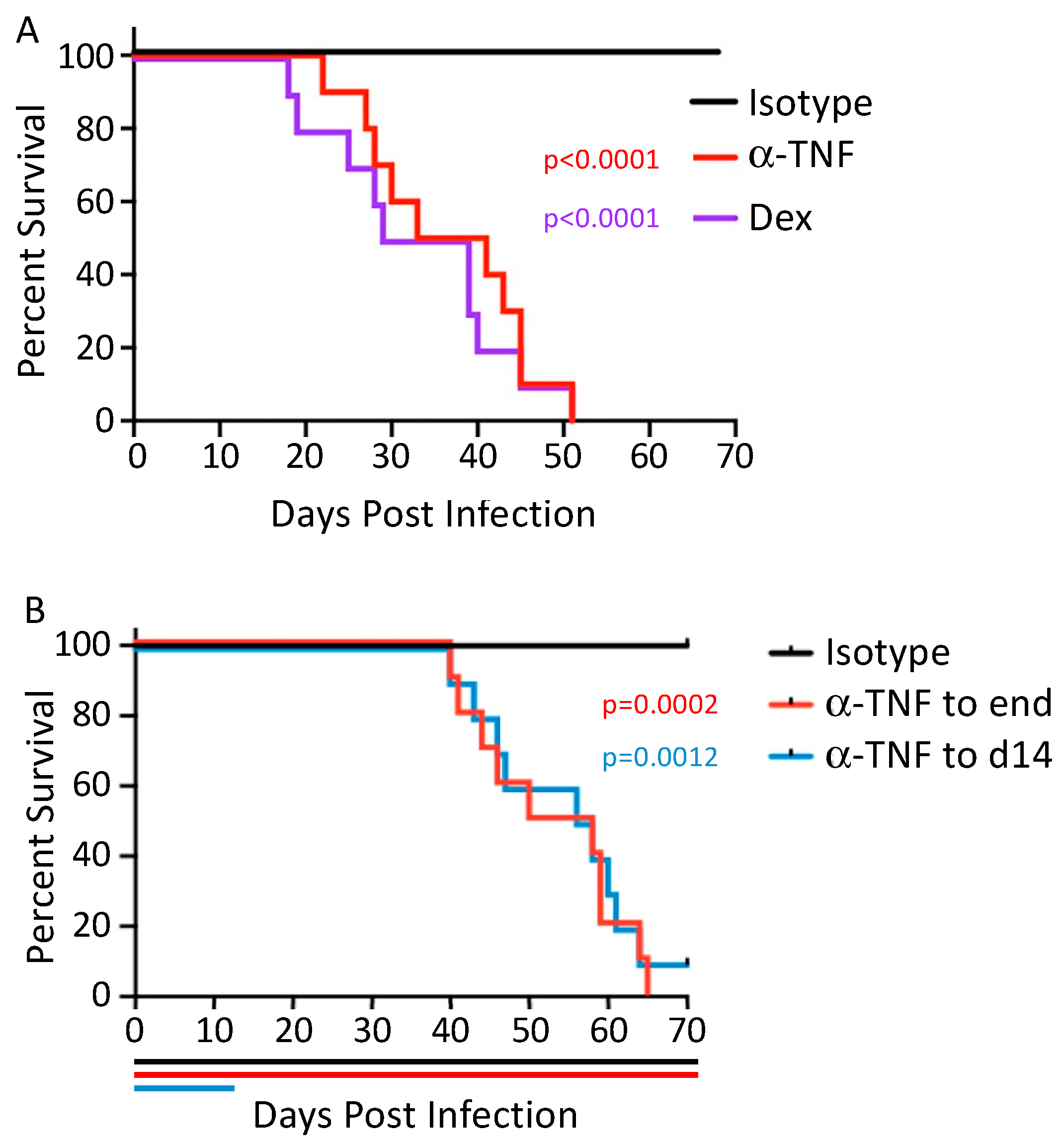
4. Centrality of TNF
5. Additional Risk Factors
6. Male Sex
7. Pregnancy
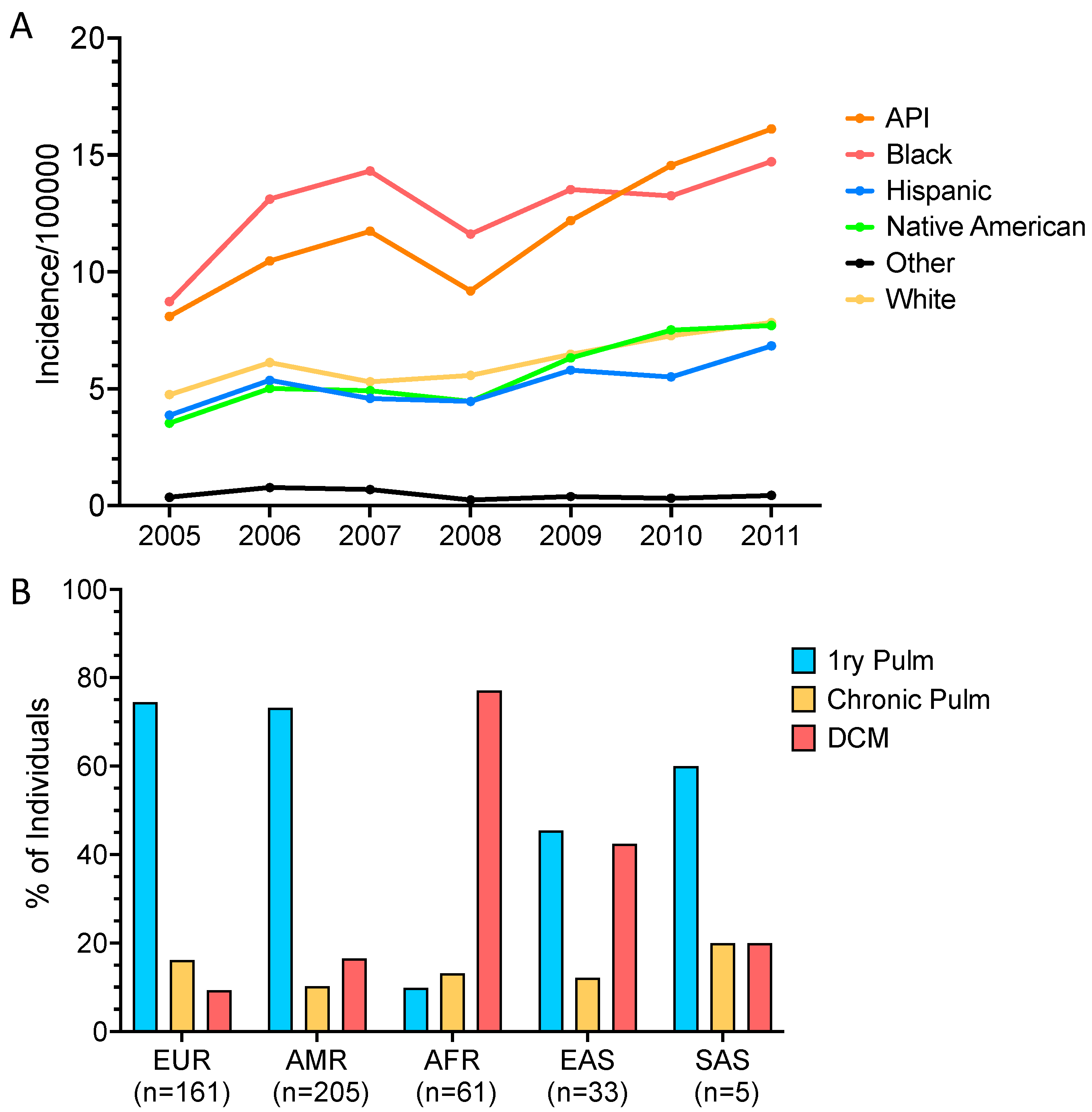
8. African American Susceptibility
9. Filipino Susceptibility
10. Erythema Nodosum and Coccidioides
11. Blood Group Association
12. Unanswered Questions
13. Conclusions
Funding
Conflicts of Interest
References
- Hung, C.Y.; Seshan, K.R.; Yu, J.J.; Schaller, R.; Xue, J.; Basrur, V.; Gardner, M.J.; Cole, G.T. A metalloproteinase of Coccidioides posadasii contributes to evasion of host detection. Infect. Immun. 2005, 73, 6689–6703. [Google Scholar] [CrossRef]
- Smith, C.E.; Beard, R.R.; Whiting, E.G.; Rosenberger, H.G. Varieties of coccidioidal infection in relation to the epidemiology and control of the diseases. Am. J. Public Health Nations Health 1946, 36, 1394–1402. [Google Scholar] [CrossRef]
- Galgiani, J.N.; Ampel, N.M.; Blair, J.E.; Catanzaro, A.; Geertsma, F.; Hoover, S.E.; Johnson, R.H.; Kusne, S.; Lisse, J.; MacDonald, J.D.; et al. Executive Summary: 2016 Infectious Diseases Society of American (IDSA) Clinical practice guideline for the treatment of Coccidioidomycosis. Clin. Infect. Dis. 2016, 63, 717–722. [Google Scholar] [CrossRef]
- Hsu, A.P.; Korzeniowska, A.; Aguilar, C.C.; Gu, J.; Karlins, E.; Oler, A.J.; Chen, G.; Reynoso, G.V.; Davis, J.; Chaput, A.; et al. Immunogenetics associated with severe coccidioidomycosis. JCI Insight 2022, 22, e159491. [Google Scholar] [CrossRef]
- Krogstad, P.; Thompson, G.R., 3rd; Heidari, A.; Kuran, R.; Stephens, A.V.; Butte, M.J.; Johnson, R. A clinicopathological categorization system for clinical research in coccidioidomycosis. Open Forum Infect. Dis. 2023, 10, ofad597. [Google Scholar] [CrossRef]
- Center for Disease Control. Available online: https://www.cdc.gov/fungal/fungal-disease-reporting-table.html (accessed on 2 January 2024).
- Center for Disease Control. Available online: https://www.cdc.gov/fungal/diseases/coccidioidomycosis/statistics.html (accessed on 2 January 2024).
- Tsang, C.A.; Anderson, S.M.; Imholte, S.B.; Erhart, L.M.; Chen, S.; Park, B.J.; Christ, C.; Komatsu, K.K.; Chiller, T.; Sunenshine, R.H. Enhanced surveillance of coccidioidomycosis, Arizona, USA, 2007–2008. Emerg. Infect. Dis. 2010, 16, 1738–1744. [Google Scholar] [CrossRef]
- Grizzle, A.J.; Wilson, L.; E Nix, D.; Galgiani, J.N. Clinical and economic burden of valley fever in Arizona: An incidence-based cost-of-illness analysis. Open Forum Infect. Dis. 2021, 8, ofaa623. [Google Scholar] [CrossRef]
- Wilson, L.; Ting, J.; Lin, H.; Shah, R.; MacLean, M.; Peterson, M.W.; Stockamp, N.; Libke, R.; Brown, P. The rise of valley fever: Prevalence and cost burden of coccidioidomycosis infection in California. Int. J. Environ. Res. Public Health 2019, 16, 1113. [Google Scholar] [CrossRef]
- Fish, D.G.; Ampel, N.M.; Galgiani, J.N.M.; Dols, C.L.R.; Kelly, P.C.M.; Johnson, C.H.M.; Pappagianis, D.M.; Edwards, J.E.M.; Wasserman, R.B.M.; Clark, R.J.M.; et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine 1990, 69, 384–391. [Google Scholar] [CrossRef]
- Buchacz, K.; Lau, B.; Jing, Y.; Abraham, A.G.; Gill, M.J.; Silverberg, M.J.; Goedert, J.J.; Sterling, T.R.; Althoff, K.N.; Martin, J.N.; et al. Incidence of AIDS-defining opportunistic infections in a multicohort analysis of HIV-infected persons in the United States and Canada, 2000–2010. J. Infect. Dis. 2016, 214, 862–872. [Google Scholar] [CrossRef]
- Blair, J.E.; Logan, J.L. Coccidioidomycosis in solid organ transplantation. Clin. Infect. Dis. 2001, 33, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, L.; Yocum, D.E.; Ampel, N.M.; Villanueva, I.; Lisse, J.; Gluck, O.; Tesser, J.; Posever, J.; Miller, M.; Araujo, J.; et al. Increased risk of coccidioidomycosis in patients treated with tumor necrosis factor α antagonists. Arthritis Rheum. 2004, 50, 1959–1966. [Google Scholar] [CrossRef]
- Donovan, F.M.; Ramadan, F.A.; Lim, J.R.; Buchfuhrer, J.E.; Khan, R.N.; DeQuillfeldt, N.P.; Davis, N.M.; Kaveti, A.; De Shadarevian, M.; Bedrick, E.J.; et al. Contribution of biologic response modifiers to the risk of Coccidioidomycosis severity. Open Forum Infect. Dis. 2022, 9, ofac032. [Google Scholar] [CrossRef]
- Sondermeyer Cooksey, G.L.; Nguyen, A.; Vugia, D.; Jain, S. Regional Analysis of Coccidioidomycosis Incidence—California, 2000–2018. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1817–1821. [Google Scholar] [CrossRef]
- Mccurdy, S.A.; Portillo-Silva, C.; Sipan, C.L.; Bang, H.; Emery, K.W. Risk for Coccidioidomycosis among Hispanic farm workers, California, USA, 2018. Emerg. Infect. Dis. 2020, 26, 1430–1437. [Google Scholar] [CrossRef]
- de Perio, M.A.; Materna, B.L.; Sondermeyer Cooksey, G.L.; Vugia, D.J.; Su, C.-P.; Luckhaupt, S.E.; McNary, J.; Wilken, J.A. Occupational coccidioidomycosis surveillance and recent outbreaks in California. Med. Mycol. 2019, 57, S41–S45. [Google Scholar] [CrossRef]
- Bronnimann, D.A.; Adam, R.D.; Galgiani, J.N.; Habib, M.P.; Petersen, E.A.; Porter, B.; Bloom, J.W. Coccidioidomycosis in the acquired immunodeficiency syndrome. Ann. Intern. Med. 1987, 106, 372–379. [Google Scholar] [CrossRef]
- Ampel, N.M.; Dols, C.L.; Galgiani, J.N. Coccidioidomycosis during human immunodeficiency virus infection: Results of a prospective study in a coccidioidal endemic area. Am. J. Med. 1993, 94, 235–240. [Google Scholar] [CrossRef]
- Woods, C.W.; McRill, C.; Plikaytis, B.D.; Rosenstein, N.E.; Mosley, D.; Boyd, D.; England, B.; Perkins, B.A.; Ampel, N.M.; Hajjeh, R.A. Coccidioidomycosis in human immunodeficiency virus–infected persons in Arizona, 1994–1997: Incidence, risk factors, and prevention. J. Infect. Dis. 2000, 181, 1428–1434. [Google Scholar] [CrossRef]
- Masannat, F.Y.; Ampel, N.M. Coccidioidomycosis in patients with HIV-1 infection in the era of potent antiretroviral therapy. Clin. Infect. Dis. 2010, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.K.; Galgiani, J.N.; O’Donnell, M.R.; Ito, J.I.; Beatty, P.G.; Evans, T.G. Coccidioidomycosis in bone marrow transplant recipients. Transplantation 1993, 56, 1531–1533. [Google Scholar]
- Glenn, T.J.; Blair, J.E.; Adams, R.H. Coccidioidomycosis in hematopoietic stem cell transplant recipients. Med. Mycol. 2005, 43, 705–710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schriber, J.; Almond, H.; Alvarnas, J.; Sarkadee-Adoo, C. Successful allogeneic bone marrow transplantation in a patient with active Coccidioidomycosis. Bone Marrow Transplant. 2005, 35, 927–928. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.A.; Kuo, P.H.; Yeager, A.M. Disseminated coccidioidomycosis masquerading as recurrent lymphoma. BMJ Case Rep. 2018, 2018, bcr2018224965. [Google Scholar] [CrossRef] [PubMed]
- Multani, A.; Allard, L.S.; Wangjam, T.; Sica, R.A.; Epstein, D.J.; Rezvani, A.R.; Ho, D.Y. Missed diagnosis and misdiagnosis of infectious diseases in hematopoietic cell transplant recipients: An autopsy study. Blood Adv. 2019, 3, 3602–3612. [Google Scholar] [CrossRef]
- Saling, C.F.; Gea-Banacloche, J.; Trickett, J.S.; Blair, J.E. Coccidioidomycosis in allogeneic stem cell transplant recipients: Case series and review of the literature. J. Fungi 2021, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.L.R.; Razonable, R.R. A review of endemic mycoses after hematopoietic stem cell transplantation. Transpl. Infect. Dis. 2023, 25, e14155. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, D.; Carey, E.J.; Blair, J.E. Coccidioidomycosis in liver transplant recipients in an endemic area. Am. J. Transplant. 2011, 11, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.; Kelley, K.D.; Dandekar, S.; Cohen, S.H.; Thompson, G.R., 3rd. Case Series of End-Stage Liver Disease Patients with Severe Coccidioidomycosis. J. Fungi 2023, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Blohm, J.E.; Panthula, M.; Aggarwal, A.; Swazo, R.; Ashoka, A.; Ainapurapu, B. Fatal Disseminated Coccidioidomycosis in Cirrhosis: A Case Series. Am. J. Med. 2023, 136, 707–709. [Google Scholar] [CrossRef]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Lohrmann, G.M.; Vucicevic, D.; Lawrence, R.; Steidley, D.E.; Scott, R.L.; Kusne, S.; Blair, J.E. Single-center experience of antifungal prophylaxis for coccidioidomycosis in heart transplant recipients within an endemic area. Transpl. Infect. Dis. 2017, 19, e12744. [Google Scholar] [CrossRef] [PubMed]
- Asbury, K.; Blair, J.E.; August, J.; Beatty, N.L.; Mi, L.; Carey, E.J.; Huskey, J.L.; LeMond, L.M.; Zangeneh, T.T. De novo coccidioidomycosis among solid organ transplant recipients 1 or more years after transplant. Am. J. Transplant. 2019, 19, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Meinke, L.; Ateeli, H.; Knox, K.S.; Raz, Y.; Ampel, N.M. Coccidioidomycosis among persons undergoing lung transplantation in the coccidioidal endemic region. Transpl. Infect. Dis. 2017, 19, e12713. [Google Scholar] [CrossRef] [PubMed]
- Truong, C.N.; Nailor, M.D.; Walia, R.; Cherrier, L.; Nasar, A.; Goodlet, K.J. Universal lifelong fungal prophylaxis and risk of coccidioidomycosis in lung transplant recipients living in an endemic area. Clin. Infect. Dis. 2021, 74, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Delafield, N.L.; Mesbah, Z.; Lacy, C.R.; Panicker, R.R.; Pasha, S.F.; Mertz, L.E.; Yiannias, J.A.; E Blair, J. Coccidioidomycosis in patients with various inflammatory disorders treated with tumor necrosis factor α inhibitors. Med. Mycol. 2021, 59, 720–727. [Google Scholar] [CrossRef]
- Taroumian, S.; Knowles, S.L.; Lisse, J.R.; Yanes, J.; Ampel, N.M.; Vaz, A.; Galgiani, J.N.; Hoover, S.E. Management of coccidioidomycosis in patients receiving biologic response modifiers or disease-modifying antirheumatic drugs. Arthritis Care Res. 2012, 64, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.E.; Ampel, N.M.; Hoover, S.E. Coccidioidomycosis in selected immunosuppressed hosts. Med. Mycol. 2019, 57, S56–S63. [Google Scholar] [CrossRef]
- Benedict, K.; McCotter, O.Z.; Brady, S.; Komatsu, K.; Sondermeyer, G.L.; Cooksey, G.L.S.; Nguyen, A.; Jain, S.; Vugia, D.J.; Jackson, B.R. Surveillance for Coccidioidomycosis–United States, 2011–2017. MMWR Surveill. Summ. 2019, 68, 1–15. [Google Scholar] [CrossRef]
- Stern, N.G.; Galgiani, J.N. Coccidioidomycosis among scholarship athletes and other college students, Arizona, USA. Emerg. Infect. Dis. 2010, 16, 321–323. [Google Scholar] [CrossRef]
- Gonzalez, R.; Naeem, F.; Ozaki, Y.; Vijayan, V. Disseminated Coccidioidomycosis in an Adolescent with Crohn’s Disease. Cureus 2021, 13, e19980. [Google Scholar] [CrossRef] [PubMed]
- Kusne, Y.; Kimes, K.E.; Feller, F.F.; Patron, R.; Banacloche, J.G.; Blair, J.E.; Vikram, H.R.; Ampel, N.M. Coccidioidomycosis in patients treated with ruxolitinib. Open Forum Infect. Dis. 2020, 7, ofaa167. [Google Scholar] [CrossRef] [PubMed]
- Odio, C.D.; Marciano, B.E.; Galgiani, J.N.; Holland, S.M. Risk factors for disseminated Coccidioidomycosis, United States. Emerg. Infect. Dis. 2017, 23, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Shubitz, L.F.; Powell, D.A.; Dial, S.M.; Butkiewicz, C.D.; Trinh, H.T.; Hsu, A.P.; Buntzman, A.; Frelinger, J.A.; Galgiani, J.N. Reactivation of Coccidioidomycosis in a mouse model of asymptomatic controlled disease. J. Fungi 2022, 8, 991. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.A.; Shubitz, L.F.; Butkiewicz, C.D.; Trinh, H.T.; Donovan, F.M.; Frelinger, J.A.; Galgiani, J.N. TNFα blockade inhibits both initial and continued control of pulmonary Coccidioides. Front. Cell. Infect. Microbiol. 2022, 11, 796114. [Google Scholar] [CrossRef]
- Powers, A.E.; Bender, J.M.; Kumánovics, A.; Ampofo, K.; Augustine, N.; Pavia, A.T.; Hill, H.R. Coccidioides immitis meningitis in a patient with hyperimmunoglobulin E syndrome due to a novel mutation in signal transducer and activator of transcription. Pediatr. Infect. Dis. J. 2009, 28, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Sugui, J.A.; Hsu, A.P.; Freeman, A.F.; Holland, S.M. Invasive fungal disease in autosomal-dominant Hyper-IgE syndrome. J. Allergy Clin. Immunol. 2010, 125, 1389–1390. [Google Scholar] [CrossRef] [PubMed]
- Odio, C.D.; Milligan, K.L.; McGowan, K.; Spergel, A.K.R.; Bishop, R.; Boris, L.; Urban, A.; Welch, P.; Heller, T.; Kleiner, D.; et al. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. J. Allergy Clin. Immunol. 2015, 136, 1411–1413.e2. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.D.; Dajud, M.V. Visual changes in a 4-year-old. Clin. Pediatr. 2008, 47, 959–961. [Google Scholar] [CrossRef]
- Vinh, D.C.; Masannat, F.; Dzioba, R.B.; Galgiani, J.N.; Holland, S.M. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-γ receptor 1 deficiency. Clin. Infect. Dis. 2009, 49, e62–e65. [Google Scholar] [CrossRef]
- Vinh, D.C.; Schwartz, B.; Hsu, A.P.; Miranda, D.J.; Valdez, P.A.; Fink, D.; Lau, K.P.; Long-Priel, D.; Kuhns, D.B.; Uzel, G.; et al. Interleukin-12 receptor β1 deficiency predisposing to disseminated coccidioidomycosis. Clin. Infect. Dis. 2011, 52, e99–e102. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.P.; Hsu, A.P.; Pechacek, J.; Bax, H.I.; Dias, D.L.; Paulson, M.L.; Chandrasekaran, P.; Rosen, L.B.; Carvalho, D.S.; Ding, L.; et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 2013, 131, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.A.; Hsu, A.P.; Shubitz, L.F.; Butkiewicz, C.D.; Moale, H.; Trinh, H.T.; Doetschman, T.; Georgieva, T.G.; Reinartz, D.M.; Wilson, J.E.; et al. Mouse model of a human STAT4 point mutation that predisposes to disseminated coccidioidomycosis. Immunohorizons 2022, 6, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Imran, T.; Cui, C. GATA2 transcription factor deficiency predisposing to severe disseminated Coccidioidomycosis. In Proceedings of the 15th International Congress of Immunology (ICI), Milan, Italy, 22–27 August 2013. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Griffin, H.; Bigley, V.; Reynard, L.N.; Hussain, R.; Haniffa, M.; Lakey, J.H.; Rahman, T.; Wang, X.-N.; McGovern, N.; et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011, 118, 2656–2658. [Google Scholar] [CrossRef]
- Perez Cavazos, S.; De la Cruz Cruz, R.A.; Castillo Bejarano, J.I.; Aparicio, D.N.V.; Mascareñas de Los Santos, A.H.; Del Carmen Zárate Hernández, M. Disseminated Coccidioidomycosis as the first presentation of a C-terminal NFKB2 pathogenic variant: A case report and review of the literature. Pediatr. Infect. Dis. J. 2022, 41, 140–144. [Google Scholar] [CrossRef]
- Chen, K.; Coonrod, E.M.; Kumánovics, A.; Franks, Z.F.; Durtschi, J.D.; Margraf, R.L.; Wu, W.; Heikal, N.M.; Augustine, N.H.; Ridge, P.G.; et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013, 93, 812–824. [Google Scholar] [CrossRef]
- Krase, I.Z.; Woodward, J.; Bauer, C.S.; Miller, H.; Sacco, K. Seronegative mediastinal coccidioidomycosis as a novel presentation of CTPS1 combined immunodeficiency. Open Forum Infect. Dis. 2022, 9, ofac403. [Google Scholar] [CrossRef]
- Viriyakosol, S.; Jimenez Mdel, P.; Gurney, M.A.; Ashbaugh, M.E.; Fierer, J. Dectin-1 is required for resistance to coccidioidomycosis in mice. mBio 2013, 4, e00597-12. [Google Scholar] [CrossRef]
- del Pilar Jiménez-A, M.; Viriyakosol, S.; Walls, L.; Datta, S.K.; Kirkland, T.; Heinsbroek, S.E.M.; Brown, G.; Fierer, J. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a). Genes Immun. 2008, 9, 338–348. [Google Scholar] [CrossRef]
- Underhill, D.M.; Rossnagle, E.; Lowell, C.A.; Simmons, R.M. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005, 106, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Witta, J.; Baffi, J.; Lekstrom, K.; Leto, T.L. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 2003, 17, 1502–1504. [Google Scholar] [CrossRef]
- Sarr, D.; Tóth, E.; Gingerich, A.; Rada, B. Antimicrobial actions of dual oxidases and lactoperoxidase. J. Microbiol. 2018, 56, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Marques, V.; Silva, T.; Cunha, F.; Covas, G.; Marinho, H.S.; Antunes, F.; Cyrne, L. A quantitative study of the cell-type specific modulation of c-Rel by hydrogen peroxide and TNF-α. Redox Biol. 2013, 1, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Everhart, M.B.; Han, W.; Sherrill, T.P.; Arutiunov, M.; Polosukhin, V.V.; Burke, J.R.; Sadikot, R.T.; Christman, J.W.; Yull, F.E.; Blackwell, T.S. Duration and intensity of NF-κB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 2006, 176, 4995–5005. [Google Scholar] [CrossRef]
- Keane, J.; Gershon, S.; Wise, R.P.; Mirabile-Levens, E.; Kasznica, J.; Schwieterman, W.D.; Siegel, J.N.; Braun, M.M. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 2001, 345, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Solovic, I.; Sester, M.; Gomez-Reino, J.J.; Rieder, H.L.; Ehlers, S.; Milburn, H.J.; Kampmann, B.; Hellmich, B.; Groves, R.; Schreiber, S.; et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: A TBNET consensus statement. Eur. Respir. J. 2010, 36, 1185–1206. [Google Scholar] [CrossRef]
- McHardy, I.; Reagan, K.L.; Sebastian, J.F.; Barker, B.; Bays, D.J.; Dandekar, S.; Cohen, S.H.; Jennings, K.E.; Sykes, J.; Thompson, G.R. Sex differences in susceptibility to Coccidioidomycosis. Open Forum Infect. Dis. 2022, 9, ofab543. [Google Scholar] [CrossRef] [PubMed]
- Crum, N.F.; Ballon-Landa, G. Coccidioidomycosis in pregnancy: Case report and review of the literature. Am. J. Med. 2006, 119, 993.e11–993.e17. [Google Scholar] [CrossRef]
- Rosenstein, N.E.; Emery, K.W.; Werner, S.B.; Kao, A.; Johnson, R.; Rogers, D.; Vugia, D.; Reingold, A.; Talbot, R.; Plikaytis, B.D.; et al. Risk factors for severe pulmonary and disseminated coccidioidomycosis: Kern County, California, 1995–1996. Clin. Infect. Dis. 2001, 32, 708–714. [Google Scholar] [CrossRef]
- Seitz, A.E.; Prevots, D.R.; Holland, S.M. Hospitalizations associated with disseminated coccidioidomycosis, Arizona and California, USA. Emerg. Infect. Dis. 2012, 18, 1476–1479. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.L.; Sable, D.L.; Mendez, P.; Smyth, L.T. Symptomatic Coccidioidomycosis following a Severe Natural Dust Storm: An Outbreak at the Naval Air Station, Lemoore, Calif. Chest 1979, 76, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Pappagianis, D.; Lindsay, S.; Beall, S.; Williams, P. Ethnic background and the clinical course of coccidioidomycosis (letter). Am. Rev. Respir. Dis. 1979, 120, 959–961. [Google Scholar]
- McCracken, B.E. Final Report of Coccidioidomycosis Research Project at Camp Roberts, Cal, 1 September 1952–15 October 1953; Surgeon’s Office 6th Army: USA, 1953. [Google Scholar]
- Deresinski, S.; Pappagianis, D.; Stevens, D.A. Association of ABO blood group and outcome of coccidioidal infection. Sabouraudia 1979, 17, 261–264. [Google Scholar] [CrossRef]
- Cohen, I.M.; Galgiani, J.N.; Potter, D.; Ogden, D.A. Coccidioidomycosis in renal replacement therapy. Arch. Intern. Med. 1982, 142, 489–494. [Google Scholar] [CrossRef]
- Louie, L.; Ng, S.; Hajjeh, R.; Johnson, R.; Vugia, D.; Werner, S.B.; Talbot, R.; Klitz, W. Influence of host genetics on the severity of coccidioidomycosis. Emerg. Infect. Dis. 1999, 5, 672–680. [Google Scholar] [CrossRef] [PubMed]
- MacNeal, W.J.; Taylor, R.M. Coccidioides immitis and coccidioidal granuloma. J. Med. Res. 1914, 30, 261–274. [Google Scholar]
- Durry, E.; Pappagianis, D.; Werner, S.; Hutwagner, L.; Sun, R.; Maurer, M.; McNeil, M.; Pinner, R. Coccidioidomycosis in Tulare County, California, 1991: Reemergence of an endemic disease. J. Med. Vet. Mycol. 1997, 35, 321–326. [Google Scholar] [CrossRef][Green Version]
- Bays, D.J.; Thompson, G.R.; Reef, S.; Snyder, L.; Freifeld, A.J.; Huppert, M.; Salkin, D.; Wilson, M.D.; Galgiani, J.N. Natural History of Disseminated Coccidioidomycosis: Examination of the Veterans Affairs–Armed Forces Database. Clin. Infect. Dis. 2021, 73, e3814–e3819. [Google Scholar] [CrossRef]
- Bercovitch, R.S.; Catanzaro, A.; Schwartz, B.S.; Pappagianis, D.; Watts, D.; Ampel, N.M. Coccidioidomycosis during pregnancy: A review and recommendations for management. Clin. Infect. Dis. 2011, 53, 363–368. [Google Scholar] [CrossRef]
- Singh, N.; Perfect, J.R. Immune reconstitution syndrome and exacerbation of infections after pregnancy. Clin. Infect. Dis. 2007, 45, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.A.M.; Ket, J.C.F.; Caanen, M.R.; Lambalk, C.B. Reproductive hormone concentrations in pregnancy and neonates: A systematic review. Reprod. Biomed. Online 2013, 27, 33–63. [Google Scholar] [CrossRef] [PubMed]
- Makieva, S.; Saunders, P.T.K.; Norman, J.E. Androgens in pregnancy: Roles in parturition. Hum. Reprod. Update 2014, 20, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Abigail, A.; Mika, W.; Debika, B.; Mae, Z.; Lindsay, K. Disseminated peritoneal coccidioidomycosis in pregnancy following fertility treatment: A case report and literature review. Case Rep. Women’s Health 2021, 30, e00299. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, R.; Chu, K.; Ramasamy, R. Novel Therapy for Male Hypogonadism. Curr. Urol. Rep. 2018, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Willett, F.M.; Weiss, A. Coccidioidomycosis in Southern California: Report of a new endemic area with a review of 100 cases. Ann. Intern. Med. 1945, 23, 349–375. [Google Scholar] [CrossRef]
- Flynn, N.M.; Hoeprich, P.D.; Kawachi, M.M.; Lee, K.K.; Lawrence, R.M.; Goldstein, E.; Jordan, G.W.; Kundargi, R.S.; Wong, G.A. An unusual outbreak of windborne coccidioidomycosis. N. Engl. J. Med. 1979, 301, 358–361. [Google Scholar] [CrossRef]
- Kupferwasser, D.; Miller, L.G. Sociodemographic factors associated with patients hospitalised for coccidioidomycosis in California and Arizona, State Inpatient Database 2005–2011. Epidemiol. Infect. 2021, 149, e127. [Google Scholar] [CrossRef] [PubMed]
- Gifford, M.A.; Coccidioidomycosis, Kern County. Presented Sixth Pacific Science Congress of the Pacific Science Association. 2 August 1939. Available online: https://kernpublichealth.com/wp-content/uploads/LibraryCocci-Article-1939.pdf (accessed on 21 January 2024).
- Pappagianis, D.; Einstein, H. Tempest from Tehachapi takes toll or Coccidioides conveyed aloft and afar. West. J. Med. 1978, 129, 527–530. [Google Scholar]
- Arsura, E.L.; Kilgore, W.B.; Ratnayake, S.N. Erythema Nodosum in pregnant patients with Coccidioidomycosis. Clin. Infect. Dis. 1998, 27, 1201–1203. [Google Scholar] [CrossRef][Green Version]
- Jenks, J.D.; Aneke, C.I.; Al-Obaidi, M.M.; Egger, M.; Garcia, L.; Gaines, T.; Hoenigl, M.; Thompson, G.R. Race and ethnicity: Risk factors for fungal infections? PLoS Pathog. 2023, 19, e1011025. [Google Scholar] [CrossRef] [PubMed]
- Sangha, A.M. Dermatological Conditions in SKIN OF COLOR-: Managing Atopic dermatitis. J. Clin. Aesthetic Dermatol. 2021, 14, S20–S22. [Google Scholar]
- DiCaudo, D.J.; Yiannias, J.A.; Laman, S.D.; Warschaw, K.E. The exanthem of acute pulmonary coccidioidomycosis. Clinical and histopathologic features of 3 cases and review of the literature. Arch. Dermatol. 2006, 142, 744–746. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blair, J.E.; Coakley, B.; Santelli, A.C.; Hentz, J.G.; Wengenack, N.L. Serologic testing for symptomatic coccidioidomycosis in immunocompetent and immunosuppressed hosts. Mycopathologia 2006, 162, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Nesbit, L.; Johnson, S.M.; Pappagianis, D.; Ampel, N.M. Polyfunctional T lymphocytes are in the peripheral blood of donors naturally immune to coccidioidomycosis and are not induced by dendritic cells. Infect. Immun. 2010, 78, 309–315. [Google Scholar] [CrossRef]
- Thompson, G.R., III; Coccidioidomycosis and Valley Fever. 16 December 2022. Available online: https://emedicine.medscape.com/article/215978 (accessed on 5 January 2024).
- Centers for Disease Control and Prevention (CDC). Outbreak of erythema nodosum of unknown cause–New Mexico, November 2007–January 2008. MMWR Morb. Mortal. Wkly. Rep. 2009, 58, 1347–1351. [Google Scholar]
- Pu, J.; Miranda, V.; Minior, D.; Reynolds, S.; Rayhorn, B.; Ellingson, K.D.; Galgiani, J.N. Improving early recognition of coccidioidomycosis in urgent care clinics: Analysis of an implemented education program. Open Forum Infect. Dis. 2023, 10, ofac654. [Google Scholar] [CrossRef] [PubMed]
- Petkus, A.F.; Baum, L.L. Natural killer cell inhibition of young spherules and endospores of Coccidioides immitis. J. Immunol. 1987, 139, 3107–3111. [Google Scholar] [CrossRef] [PubMed]
- Hampton, H.R.; Chtanova, T. Lymphatic migration of immune cells. Front. Immunol. 2019, 10, 1168. [Google Scholar] [CrossRef]
- Li, L.; Schmelz, M.; Kellner, E.M.; Galgiani, J.N.; Orbach, M.J. Nuclear labeling of Coccidioides posadasii with green fluorescent protein. Ann. N. Y. Acad. Sci. 2007, 1111, 198–207. [Google Scholar] [CrossRef]
- Kirkland, T.N.; Stevens, D.A.; Hung, C.-Y.; Beyhan, S.; Taylor, J.W.; Shubitz, L.F.; Duttke, S.H.; Heidari, A.; Johnson, R.H.; Deresinski, S.C.; et al. Coccidioides species: A review of basic research: 2022. J. Fungi 2022, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, O.; Houri-Ze’evi, L.; Anava, S.; Goh, W.S.S.; Kerk, S.Y.; Hannon, G.J.; Hobert, O. Starvation-induced transgenerational inheritance of small RNAs in C. elegans. Cell 2014, 158, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Trangucci, R.; Nelson, K.N.; Fu, W.; Collender, P.A.; Head, J.R.; Hoover, C.M.; Skaff, N.K.; Li, T.; Li, X.; et al. Prenatal and early-life exposure to the Great Chinese Famine increased the risk of tuberculosis in adulthood across two generations. Proc. Natl. Acad. Sci. USA 2020, 117, 27549–27555. [Google Scholar] [CrossRef]
- Johnson, R.; Ding, Y.; Venkateswaran, V.; Bhattacharya, A.; Boulier, K.; Chiu, A.; Knyazev, S.; Schwarz, T.; Freund, M.; Zhan, L.; et al. Leveraging genomic diversity for discovery in an electronic health record linked biobank: The UCLA ATLAS Community Health Initiative. Genome Med. 2022, 14, 104. [Google Scholar] [CrossRef]
- Banda, Y.; Kvale, M.N.; Hoffmann, T.J.; Hesselson, S.E.; Ranatunga, D.; Tang, H.; Sabatti, C.; Croen, L.A.; Dispensa, B.P.; Henderson, M.; et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics 2015, 200, 1285–1295. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV+ Cases | Active/Positive # | Diffuse Pulmonary Infiltrate (n) | DCM | Death | CD4 < 250/µL (n) | Reference |
|---|---|---|---|---|---|---|
| 27 | 7 | 6 | 5 | 7 | 4 (5) | [19] |
| 77 | 77 | 31 | 29 * | 32 | 46 (55) | [11] |
| 170 | 13 | 5 | 1 | 5 | 12 (13) | [20] |
| 153 | 153 | 80 (146) | 43 | 29 | 135 & (153) | [21] |
| 257 | 29 | 4 (15) | 2 | nd | 4 (9) | [22] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, A.P. The Known and Unknown “Knowns” of Human Susceptibility to Coccidioidomycosis. J. Fungi 2024, 10, 256. https://doi.org/10.3390/jof10040256
Hsu AP. The Known and Unknown “Knowns” of Human Susceptibility to Coccidioidomycosis. Journal of Fungi. 2024; 10(4):256. https://doi.org/10.3390/jof10040256
Chicago/Turabian StyleHsu, Amy P. 2024. "The Known and Unknown “Knowns” of Human Susceptibility to Coccidioidomycosis" Journal of Fungi 10, no. 4: 256. https://doi.org/10.3390/jof10040256
APA StyleHsu, A. P. (2024). The Known and Unknown “Knowns” of Human Susceptibility to Coccidioidomycosis. Journal of Fungi, 10(4), 256. https://doi.org/10.3390/jof10040256