
Screening of the Candidate Metabolite to Evaluate the Mycelium Physiological Maturation of Lyophyllum decastes Based on Metabolome and Transcriptome Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Strain and Cultivation Conditions
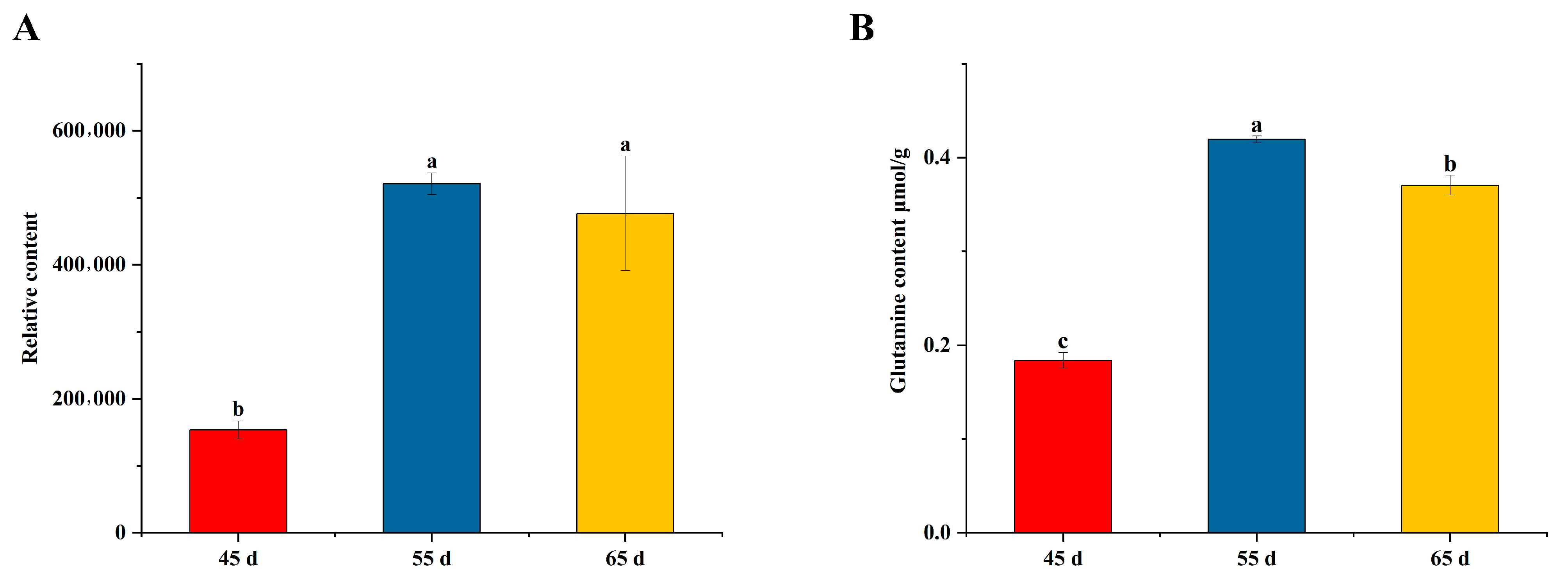
2.2. Determination of Flavonoids and Amino Acid Content
2.3. Metabolite Extraction
2.4. UPLC–MS/MS Conditions
2.5. Analyses of Qualitative and Quantitative Metabolite
2.6. RNA Extraction and RNA–Seq
2.7. Sequence Data Processing
2.8. Quantitative Real–Time PCR
2.9. Detection of Glutamine Content
3. Results
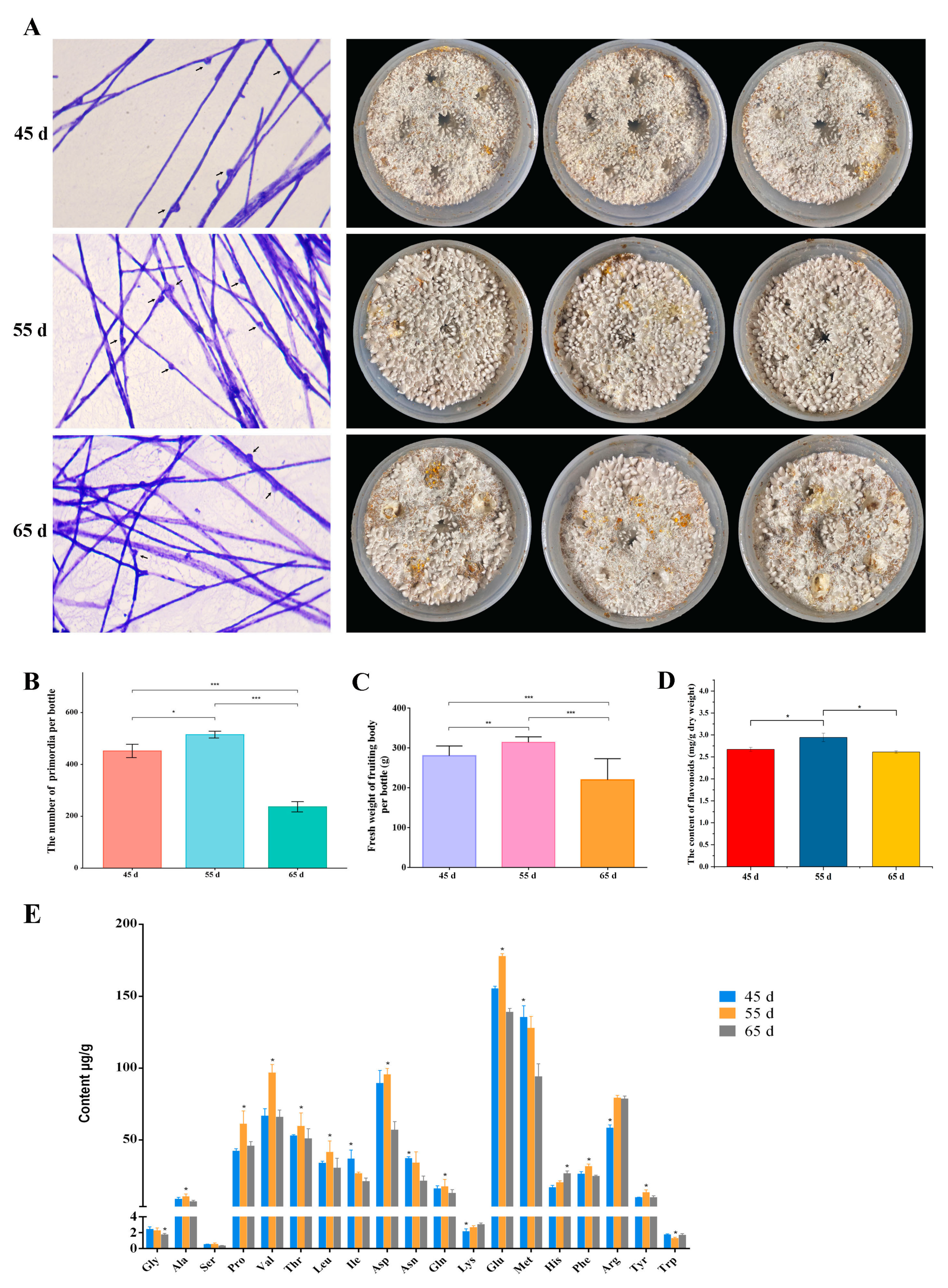
3.1. The Yield and Quality of L. decastes
3.2. The Metabolome Profiling of L. decastes
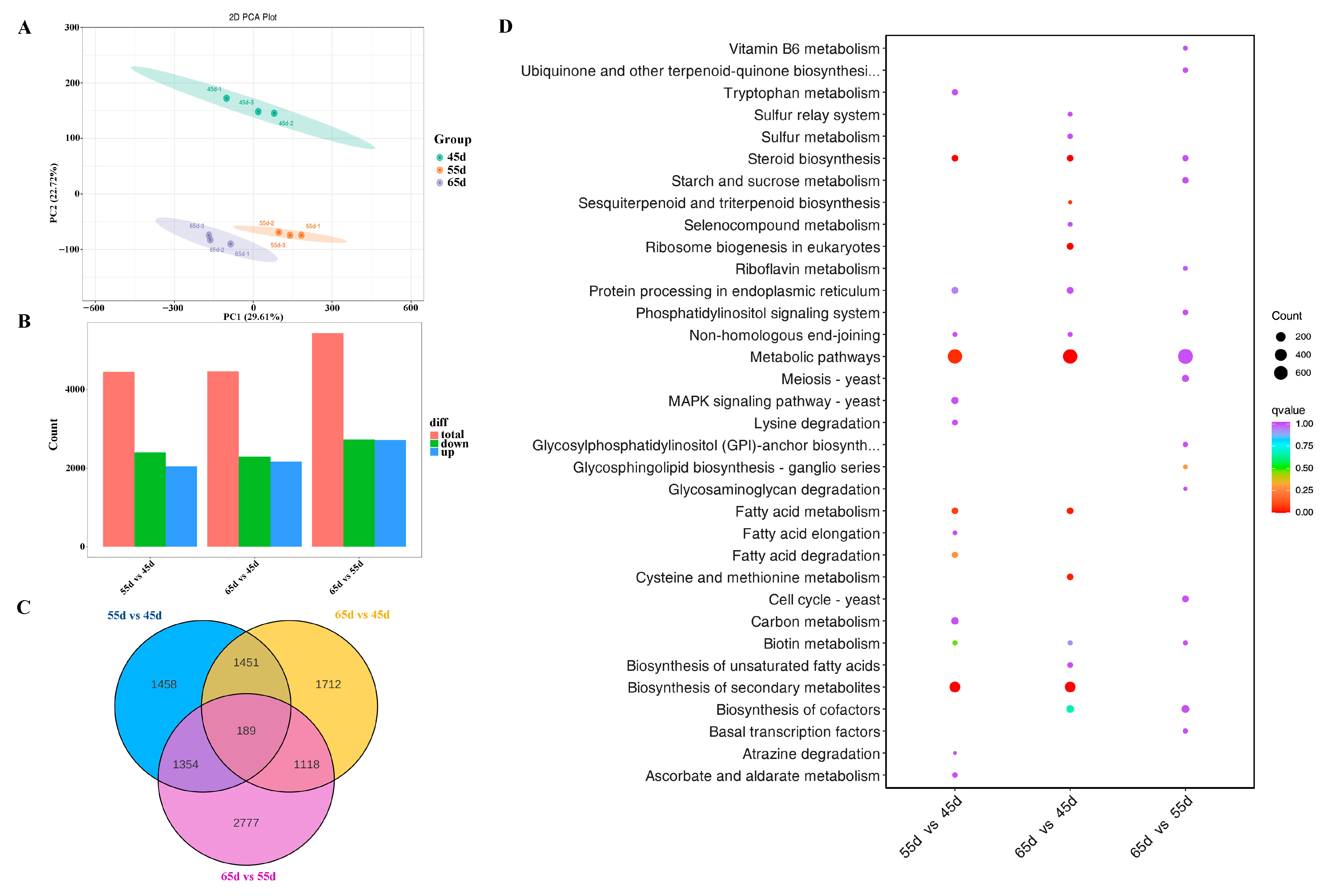
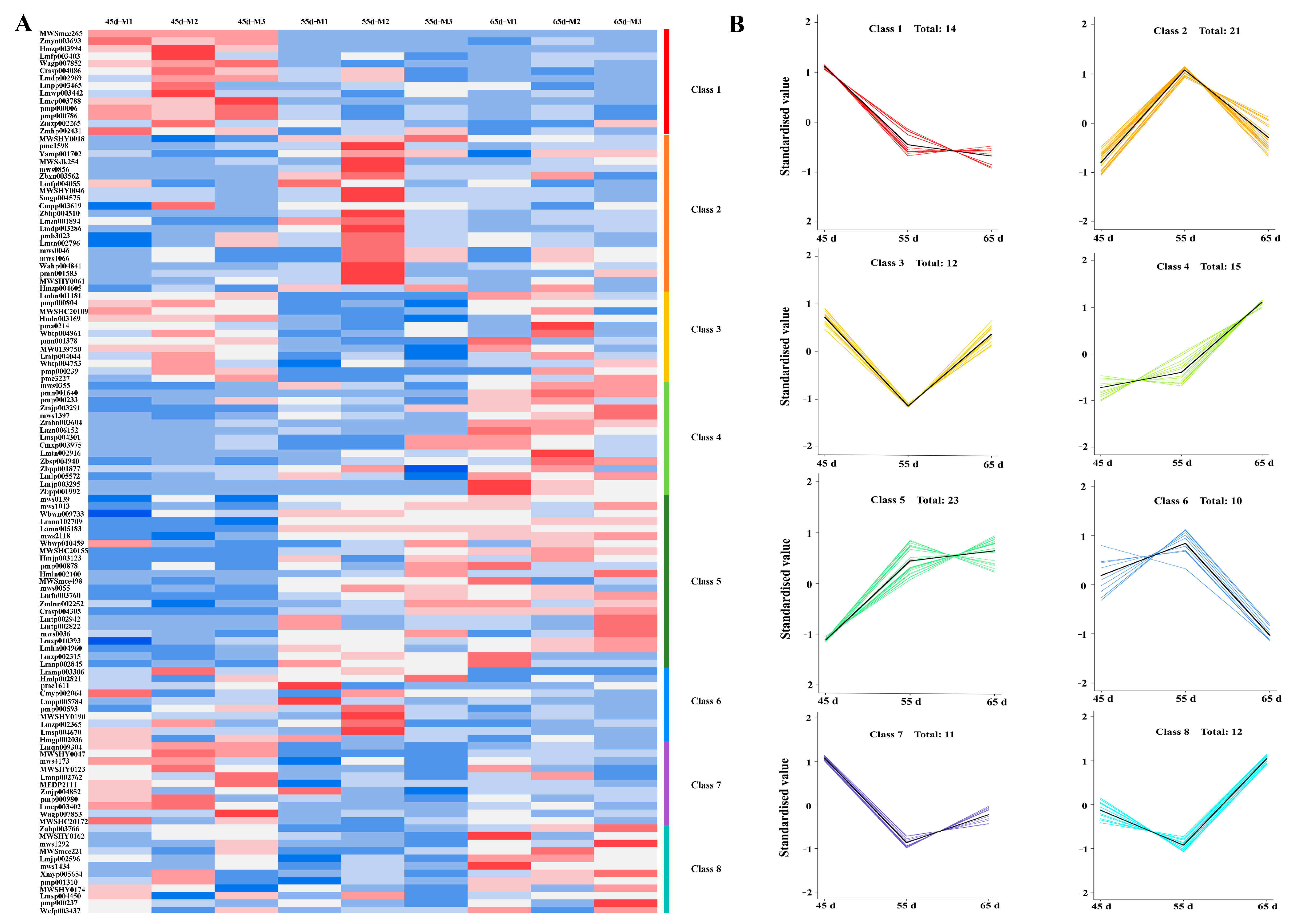
3.3. The Transcriptome Profiling of L. decastes
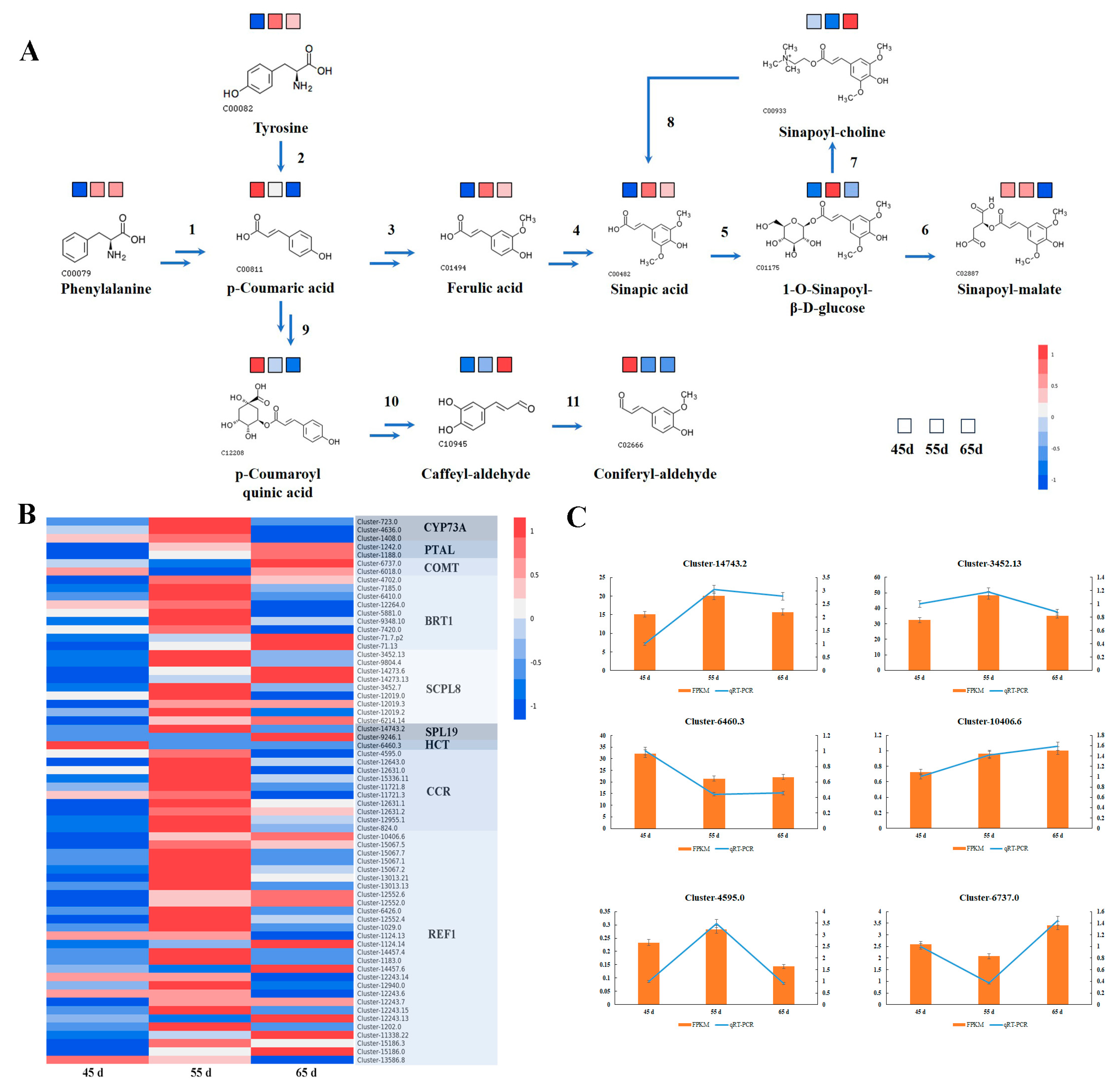
3.4. Biosynthesis of Secondary Metabolites in L. decastes
3.5. Biosynthesis of Amino Acids in L. decastes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, Y.; Li, J.; Lin, H.; Liu, P.; Zhang, F.; Lin, X.; Liang, J.; Tao, Y.; Jiang, Y.; Chen, B. Ultrasonic treatment decreases Lyophyllum decastes fruiting body browning and affects energy metabolism. Ultrason. Sonochem. 2022, 89, 106111. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Bai, H.Y.; Fu, W.Q.; Sun, K.; Wang, H.W.; Xu, D.L.; Dai, C.C.; Jia, Y. Endophytic bacteria promote the quality of Lyophyllum decastes by improving non–volatile taste components of mycelia. Food Chem. 2021, 336, 127672. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Y.; Qin, C.; Ye, S.; Zhang, F.; Linhardt, R.J.; Zhang, A. Structural characterization of an antioxidant polysaccharide isolated from the fruiting bodies of Lyophyllum decastes. J. Mol. Struct. 2023, 1285, 135507. [Google Scholar] [CrossRef]
- Miura, T.; Kubo, M.; Itoh, Y.; Iwamoto, N.; Kato, M.; Park, S.; Suzuki, I. Antidiabetic activity of Lyophyllum decastes in genetically type 2 diabetic mice. Biol. Pharm. Bull. 2002, 25, 1234–1237. [Google Scholar] [CrossRef]
- Ukawa, Y.; Furuichi, Y.; Kokean, Y.; Nishii, T.; Hisamatsu, M. Effect of hatakeshimeji (Lyophyllum decastes Sing.) mushroom on serum lipid levels in rats. J. Nutr. Sci. Vitaminol. 2002, 48, 73–76. [Google Scholar] [CrossRef]
- Ukawa, Y.; Izumi, Y.; Ohbuchi, T.; Takahashi, T.; Ikemizu, S.; Kojima, Y. Oral administration of the extract from hatakeshimeji (Lyophyllum decastes sing.) mushroom inhibits the development of atopic dermatitis–like skin lesions in NC/Nga mice. J. Nutr. Sci. Vitaminol. 2007, 53, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Ukawa, Y.; Ito, H.; Hisamatsu, M. Antitumor effects of (1→3)–beta–D–glucan and (1→6) –beta–D–glucan purified from newly cultivated mushroom, hatakeshimeji (Lyophyllum decastes Sing.). J. Biosci. Bioeng. 2000, 90, 98–104. [Google Scholar] [CrossRef]
- Xu, L.; Yang, W.; Qiu, T.; Gao, X.; Zhang, H.; Zhang, S.; Cui, H.; Guo, L.; Yu, H.; Yu, H. Complete genome sequences and comparative secretomic analysis for the industrially cultivated edible mushroom Lyophyllum decastes reveals insights on evolution and lignocellulose degradation potential. Front. Microbiol. 2023, 14, 1137162. [Google Scholar] [CrossRef]
- Hofstetter, V.; Clémençon, H.; Vilgalys, R.; Moncalvo, J. Phylogenetic analyses of the Lyophylleae (Agaricales, Basidiomycota) based on nuclear and mitochondrial rDNA sequences. Mycol. Res. 2002, 106, 1043–1059. [Google Scholar] [CrossRef]
- Xu, L.; Lin, R.; Li, X.; Zhang, C.; Yang, X.; Guo, L.; Yu, H.; Gao, X.; Hu, C. Comparative proteomic analyses within three developmental stages of the mushroom white Hypsizygus marmoreus. J. Fungi 2023, 9, 225. [Google Scholar] [CrossRef]
- Yoo, S.; Lee, H.Y.; Markkandan, K.; Moon, S.; Ahn, Y.J.; Ji, S.; Ko, J.; Kim, S.; Ryu, H.; Hong, C.P. Comparative transcriptome analysis identified candidate genes involved in mycelium browning in Lentinula edodes. BMC Genom. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Liang, Y.; Dai, Y.T.; Yang, C.T.; Duan, M.Z.; Zhang, Z.; Hu, S.N.; Zhang, Z.W.; Li, Y. De novo sequencing and transcriptome analysis of Pleurotus eryngii subsp. tuoliensis (Bailinggu) mycelia in response to cold stimulation. Molecules 2016, 21, 560. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Arshad, M.; Li, Y.; Zhang, H.; Gu, Y.; Yu, X.; Zhao, K.; Ma, M.; Zhang, L.; He, M.; et al. Transcriptomic profiling revealed important roles of amino acid metabolism in fruiting body formation at different ripening times in Hypsizygus marmoreus. Front. Microbiol. 2023, 14, 1169881. [Google Scholar] [CrossRef]
- Pani, B.K. Effect of age and quantity of spawn on milky mushroom production. Asian J. Exp. Biol. Sci. 2011, 2, 769–771. [Google Scholar]
- Meng, L.; Zhang, S.; Chen, B.; Bai, X.; Li, Y.; Yang, J.; Wang, W.; Li, C.; Li, Y.; Li, Z. The MADS–box transcription factor GlMADS1 regulates secondary metabolism in Ganoderma lucidum. Mycologia 2021, 113, 12–19. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J.; Zheng, S.; Wang, Z.; Xu, C.; Zhang, Q.; Wu, J.; Lou, H. Metabolite profiling and transcriptome analyses reveal novel regulatory mechanisms of melatonin biosynthesis in hickory. Hortic. Res. 2021, 8, 196. [Google Scholar] [CrossRef]
- Chukwu, S.C.; Ibeji, C.A.; Ogbu, C.; Oselebe, H.O.; Okporie, E.O.; Rafii, M.Y.; Oladosu, Y. Primordial initiation, yield and yield component traits of two genotypes of oyster mushroom (Pleurotus spp.) as affected by various rates of lime. Sci. Rep. 2022, 12, 19054. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.P.; Zhang, F.; Hu, X.; Luo, Y.; Wang, H.; Durrani, S.; Wu, F.; Li, B.; Zhou, Z.; Lu, Z.; et al. Glutamine involvement in nitrogen regulation of cellulase production in fungi. Biotechnol. Biofuels 2021, 14, 199. [Google Scholar] [CrossRef]
- Tudzynski, B. Nitrogen regulation of fungal secondary metabolism in fungi. Front. Microbiol. 2014, 5, 00656. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine–restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sun, X.; Diao, W.; Qi, X.; Bai, Y.; Yu, X.; Li, L.; Fang, H.; Chen, Z.; Liu, Q.; et al. Comparative transcriptome analysis revealed candidate genes involved in fruiting body development and sporulation in Ganoderma lucidum. Arch. Microbiol. 2022, 204, 514. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhou, R.; Lin, J.; Zang, X.; Wang, Q.; Wang, P.; Wang, L.; Li, Z.; Wang, W. Transcriptome and metabolome analyses reveal transcription factors regulating ganoderic acid biosynthesis in Ganoderma lucidum development. Front. Microbiol. 2022, 13, 956421. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Xia, E.H.; Li, M.; Cui, Y.Y.; Wang, P.M.; Zhang, J.X.; Xie, B.G.; Xu, J.P.; Yan, J.J.; Li, J.; et al. Transcriptome data reveal conserved patterns of fruiting body development and response to heat stress in the mushroom–forming fungus Flammulina filiformis. PLoS ONE 2020, 15, e0239890. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, X.; Liu, W. De novo transcriptomic analysis during Lentinula edodes fruiting body growth. Gene 2018, 641, 326–334. [Google Scholar] [CrossRef]
- Wang, J.Q.; Wen, X.F.; Yang, B.; Liu, D.; Li, X.; Geng, F. De novo transcriptome and proteome analysis of Dictyophora indusiata fruiting bodies provides insights into the changes during morphological development. Int. J. Biol. Macromol. 2020, 146, 875–886. [Google Scholar] [CrossRef]
- McCarthy, M.W.; Walsh, T.J. Amino acid metabolism and transport mechanisms as potential antifungal targets. Int. J. Mol. Sci. 2018, 19, 909. [Google Scholar] [CrossRef]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef]
- Du, F.; Zou, Y.; Hu, Q.; Jing, Y.; Yang, X. Metabolic profiling of Pleurotus tuoliensis during mycelium physiological maturation and exploration on a potential indicator of mycelial maturation. Front. Microbiol. 2019, 9, 3274. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 5′–3′ |
|---|---|
| Lyd–UBC–qPCR–F | GAGAAGGCACTTCACGGTCA |
| Lyd–UBC–qPCR–R | CCAACGTCTCGAACACTGGA |
| Lyd–Cluster–6737.0–qPCR–F | ATATGTTTGCCAGCGTCCCA |
| Lyd–Cluster–6737.0–qPCR–R | AAACGTCTTGGGCCCCTATG |
| Lyd–Cluster–3452.13–qPCR–F | ATCCCGACACAGCTCCTTTG |
| Lyd–Cluster–3452.13–qPCR–R | CTACTCCGACGGGTTGATCG |
| Lyd–Cluster–14743.2–qPCR–F | CAAGCCGCTGATCCGAAATG |
| Lyd–Cluster–14743.2–qPCR–R | CCCAAGGGGCTCTCAAGAAG |
| Lyd–Cluster–6460.3–qPCR–F | GATTCATCCGCGTTGTCTGC |
| Lyd–Cluster–6460.3–qPCR–R | TCAACGTCACGAGTTCCCAG |
| Lyd–Cluster–4595.0–qPCR–F | GGAGATTGAACCGGACAGCA |
| Lyd–Cluster–4595.0–qPCR–R | TGCTGAGAAGAAGGCATGGG |
| Lyd–Cluster–10406.6–qPCR–F | TGACCCACTCGTCGTTTCAG |
| Lyd–Cluster–10406.6–qPCR–R | CCAGTCTGCTTCTGCAGTCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, L.; Zang, X.; Zhang, P.; Sun, J.; Shi, Q.; Chang, S.; Ren, P.; Li, Z.; Meng, L. Screening of the Candidate Metabolite to Evaluate the Mycelium Physiological Maturation of Lyophyllum decastes Based on Metabolome and Transcriptome Analysis. J. Fungi 2024, 10, 734. https://doi.org/10.3390/jof10110734
Liang L, Zang X, Zhang P, Sun J, Shi Q, Chang S, Ren P, Li Z, Meng L. Screening of the Candidate Metabolite to Evaluate the Mycelium Physiological Maturation of Lyophyllum decastes Based on Metabolome and Transcriptome Analysis. Journal of Fungi. 2024; 10(11):734. https://doi.org/10.3390/jof10110734
Chicago/Turabian StyleLiang, Lidan, Xizhe Zang, Peijin Zhang, Jingwei Sun, Qingyun Shi, Siyuan Chang, Pengfei Ren, Zhuang Li, and Li Meng. 2024. "Screening of the Candidate Metabolite to Evaluate the Mycelium Physiological Maturation of Lyophyllum decastes Based on Metabolome and Transcriptome Analysis" Journal of Fungi 10, no. 11: 734. https://doi.org/10.3390/jof10110734
APA StyleLiang, L., Zang, X., Zhang, P., Sun, J., Shi, Q., Chang, S., Ren, P., Li, Z., & Meng, L. (2024). Screening of the Candidate Metabolite to Evaluate the Mycelium Physiological Maturation of Lyophyllum decastes Based on Metabolome and Transcriptome Analysis. Journal of Fungi, 10(11), 734. https://doi.org/10.3390/jof10110734