1. Introduction
Setophoma terrestris (H.N. Hansen) Gruyter (syn.
Pyrenochaeta terrestris (H.N. Hansen) Gorenz,
Phoma terrestris H.N. Hansen) is an important plant pathogenic fungus that can infect various plants, including
Allium cepa,
A. sativum,
A. porrum,
Avena sativa,
Calathea crocata,
Cucurbita maxima,
C. moschata,
Medicago sativa,
Oryza sativa,
Phaseolus lunatus,
Pisum sativum,
Solanum lycopersicum,
Triticum aestivum,
Vigna sinensis,
Zea mays [
1,
2,
3,
4,
5,
6,
7,
8]. It has been reported in countries including Argentina, Australia, Brazil, Canada, India, Japan, Mexico, Senegal, South Africa, the Netherlands, the United States, Venezuela, and Vietnam. Additionally, there have been reports in the Shandong and Jiangsu provinces of China [
9].
Onions are widely grown in China and are one of China’s important export vegetables. According to statistics, China’s total onion production in 2020 accounted for 22.69% of the world’s total production. The pink root rot of onions caused by
S. terrestris is one of the most severe diseases induced by this pathogen. Infected onion roots first show a light pink color that progresses to shades of red and purple, eventually shriveling and blackening, which results in poor growth of the onions [
3,
7]. The pinkish-red discoloration may extend into the bulb scales, and prolonged infection can lead to stunted plant growth. Typically, the infection is limited to the roots and outer bulb scales. Due to the severe threat posed by this pathogen and the significant economic losses that may be caused by its introduction into China, it has been included in the Chinese quarantine pest list by the Ministry of Agriculture and Rural Affairs of the People’s Republic of China (
http://dzs.customs.gov.cn/dzs/2746776/3699554/index.html; accessed 15 May 2021).
With increasing international trade, the possibility of the introduction and establishment of this pathogen in non-endemic areas is considerable. Early warning, rapid detection, risk analysis, and scientific control of invasive species are vital for preventing the spread of exotic organisms. Among these, the accurate identification of S. terrestris plays a key role in controlling its damage, halting its transmission and spread in a timely manner. Therefore, establishing a specific, sensitive, rapid, and effective molecular detection method for the pathogen is of great significance for facilitating rapid clearance of imported plants and plant products and early field diagnosis of diseases.
In the past, the identification of
S. terrestris was mainly based on morphological characteristics and phylogenetic analyses [
8,
10], which require a long time and well-trained staff. As an alternative approach, a recently developed real-time PCR approach provides fast and accurate results as compared to culture-based methods [
11]. However, real-time PCR requires high technical expertise from operators and expensive laboratory equipment, making on-site testing challenging. In comparison, recombinase polymerase amplification (RPA), a nucleic acid assay considered to be an alternative to PCR, can be performed at a constant temperature of 37–42 °C, and the time can be controlled to less than 30 min. Therefore, RPA is a suitable amplification method for portable rapid nucleic acid detection on-site.
Furthermore, the combined utilization of RPA with clustered regularly interspaced short palindromic repeats (CRISPR)-based detection technology demonstrates superior accuracy, rapidity, sensitivity, and convenience in the identification of various pathogenic microbes when compared to traditional methodologies [
12]. The foundation of this detection approach lies in the collateral nuclease activity of CRISPR-associated proteins such as Cas12 and Cas13, which recognize target nucleic acid sequences while simultaneously cleaving single-stranded DNA (ssDNA) reporter probes [
13]. Specifically, the CRISPR/Cas12a system, guided by a 41 to 44 nt single CRISPR RNA (crRNA), exhibits precise recognition and cleavage of double-stranded DNA, accompanied by the collateral cleavage of a reporter probe, thus representing a novel generation of rapid and accurate nucleotide acid detection techniques [
14]. Additionally, two prevalent signal reporter systems frequently integrated into CRISPR/Cas12a-based methodologies are paper-based lateral flow strips (PLFS) and fluorescence-based detection systems (FRB). The PLFS detection mechanism, complemented by colloidal gold nanoparticles, delivers expeditious and straightforward results within a 10 min timeframe for positive reactions, while the FRB detection method generates detectable fluorescence visible to the naked eye under blue light. This innovative platform has been effectively utilized across a few pathogenic fungi, such as diagnosing
Diaporthe aspalathi,
Diaporthe caulivora,
Fusarium verticillioides through RPA-CRISPR/Cas12a combined with a lateral flow assay [
15,
16], and detecting
Verticillium dahliae by uniting RPA-CRISPR/Cas12a with fluorescence-based detection systems [
17].
In this study, we introduce a method for the rapid on-site detection of S. terrestris using the CRISPR/Cas12a system. This strategy entails a combination of RPA reaction, Cas12a cleavage post-recognition of the target, and visualization through PLFS and FRB.
2. Materials and Methods
2.1. Sample Collection
The DNA of two
S. terrestris strains, CBS 335.29 and CBS 122483, was obtained from the culture collection (CBS) of the Westerdijk Fungal Biodiversity Institute in the Netherlands and used for molecular diagnosis. Seven closely related strains of
Setophoma were accumulated from our previous work (
Table 1), which were preserved in the LC culture collection (a personal culture collection of Lei Cai, housed in the Institute of Microbiology, Chinese Academy of Sciences). All strains used in this study were accurately identified based on morphological characteristics and multi-locus (ITS,
gapdh,
tef-1α,
tub2) phylogenetic analysis in previous studies [
8,
10,
18].
2.2. DNA Extraction
The strains were grown at 25 °C for 7 days, and the hyphae were scraped with a scalpel into the 2 mL centrifuge tube. For routine genomic DNA extraction, we used a modified CTAB protocol [
19]. DNA concentration was quantified using NanoDrop™One (Thermo Fisher Scientific, Waltham, MA, USA).
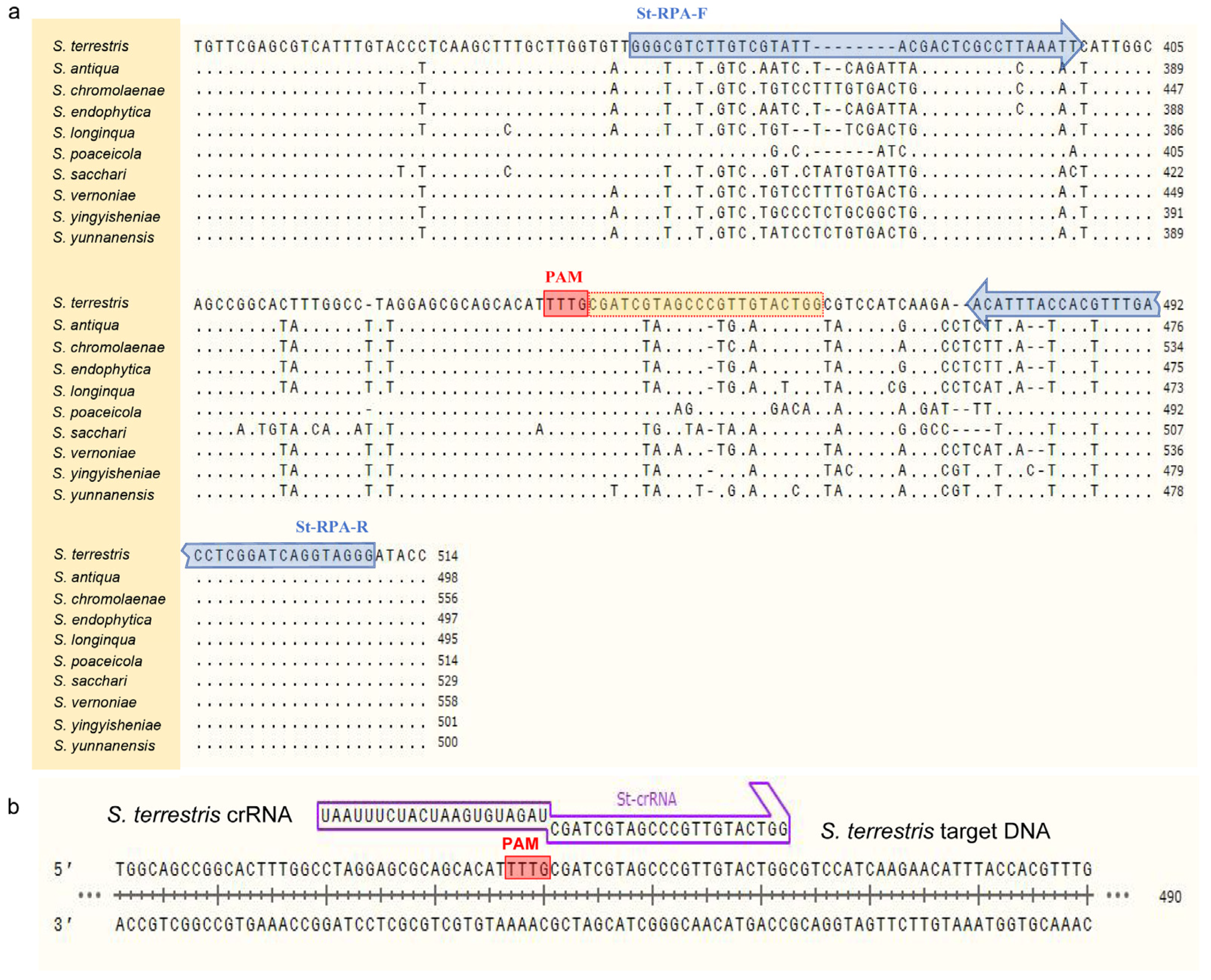
2.3. Design of Primers and crRNA
ITS sequences of
Setophoma strains listed in
Table 1 were obtained from the NCBI GenBank and aligned using MAFFT v. 7 “
http://mafft.cbrc.jp/alignment/server/index.html (accessed on 10 May 2023)”. After that, the conservative region of
S. terrestris and the variant region for other species were chosen to design primer pairs St-RPA-F/St-RPA-R according to the manual of TwistAmp
® DNA Amplification Kits (
Figure 1 and
Table 2). It is worth noting that, when determining target sequences and designing primers, the amplicon contained at least one protospacer adjacent motif (PAM) site (5′-TTTN-3′) to facilitate recognition by Cas12a [
14].
For Cas12a cleavage, the crRNA used in the CRISPR detection system was designed based on the amplified product from
S. terrestris (
Figure 1) and synthesized by Sangon Biotech. The FAM/BHQ1 labeled single-stranded DNA (ssDNA) was employed and cleaved for the RPA-Cas12a-mediated real-time and end-point fluorescence assay. In contrast, the FAM/Biotin labeled ssDNA was employed for lateral flow strip detection. The complete sequence of crRNA included a direct repeat sequence for Cas12a recognition 5′-UAAUUUCUACUAAGUGUAGAU-3′ (scaffold sequence) and a spacer sequence 5′-CGAUCGUAGCCCGUUGUACUGG-3′ (guide sequence) (
Figure 1).
2.4. RPA Reaction
The RPA reaction was prepared using the TwistAmp™ Basic Kit (TwistDx™, Cambridge, UK). The RPA was generated by adding 29.5 μL rehydration buffer, 11.2 μL DEPC-H2O, 2.4 μL St-RPA-F primer (10 μmol/L), 2.4 μL St-RPA-R primer (10 μmol/L), 2 μL extracted DNA, and 2.5 μL magnesium acetate (280 mmol/L) to preconfigured reaction tubes containing the recombinase, polymerase, and single-strand binding protein. The mixture was then incubated at 39 °C for 30 min using a T30D tri-block super-gradient PCR system (LongGene, Hangzhou, Zhejiang, China). Then, the amplification products were detected by agarose gel electrophoresis.
To optimize the RPA reaction condition, the other parameters were kept unchanged, and the RPA time (set to 10, 20, 25, 30, 40 min) was adjusted to select the optimal reaction time based on the band brightness. After that, using the selected time and keeping other parameters unchanged, RPA temperature (set to 33, 35, 37, 39 and 41 °C) was adjusted to select the optimized temperature based on the band brightness.
2.5. CRISPR/Cas12a-FRB Visual Detection
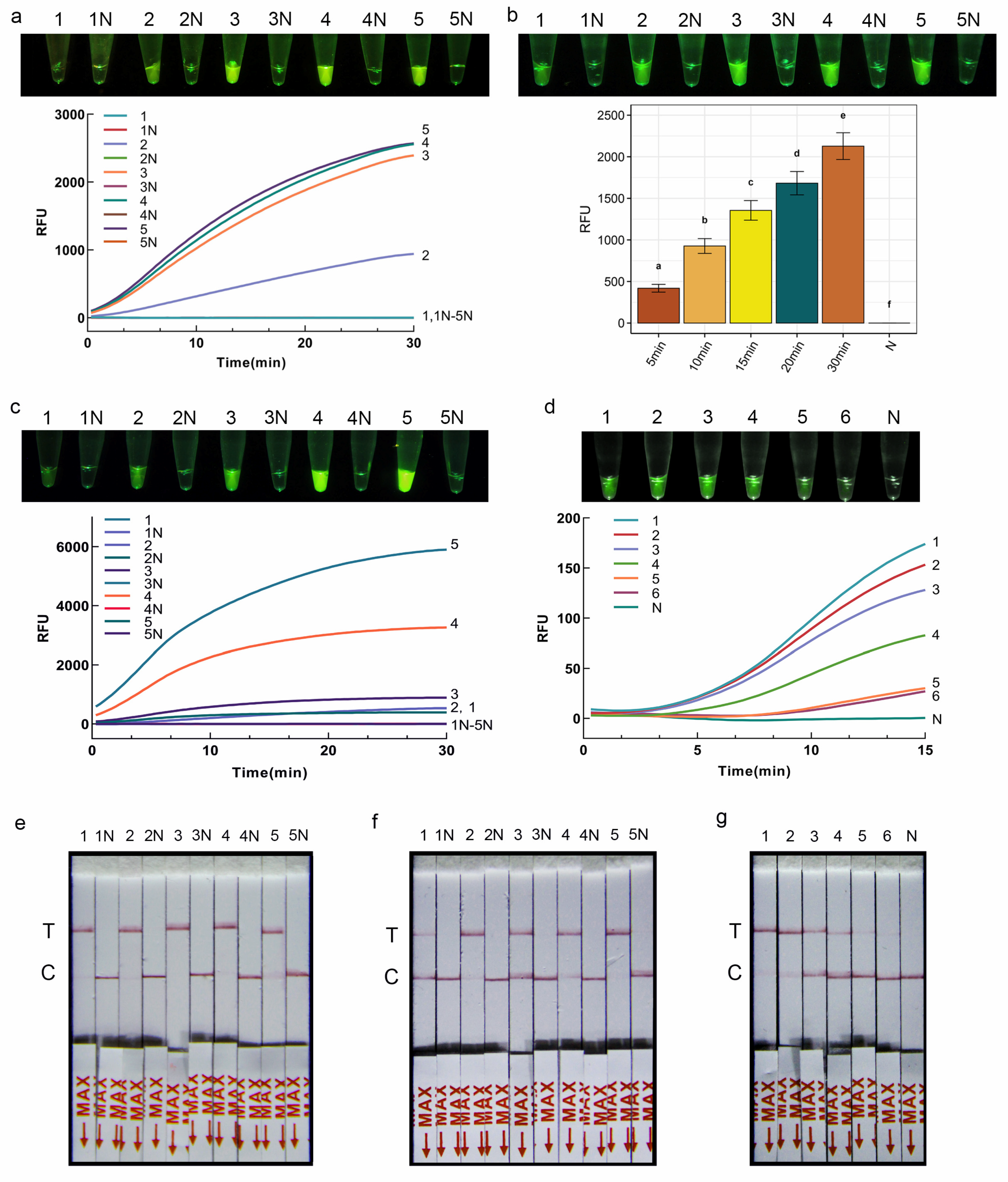
The RPA-CRISPR/Cas12a-FRB assay system contains 2 μL of the above RPA product, 14.4 μL DEPC-H2O, 2 μL NEBuffer (10×), 0.4 μL Cas12a (5 μmol/L, New England Biolabs, Ipswich, MA, USA), 0.4 μL St-crRNA (0.01 mmol/L), 0.8 μL FQ-DNA reporter (5 μmol/L). After incubating under 37 °C for 30 min to perform CRISPR/Cas12a cleavage assay, the products were detected directly by the naked eye under blue light. Positive reactions were indicated by visible green fluorescent light, whereas negative reaction systems were transparent and colorless.
Next, to optimize the reaction conditions of the CRISPR/Cas12a-FRB detection system, the concentration ratio of Cas12a to crRNA was systematically adjusted (0 nM/0 nM, 50 nM/100 nM, 100 nM/200 nM, 150 nM/250 nM, 200 nM/300 nM), with subsequent observation of the fluorescence signal intensity under each condition. Subsequently, the concentration of the fluorescent reporter was fine-tuned, with the FQ-DNA concentrations set at 50 nmol/L, 100 nmol/L, 200 nmol/L, 400 nmol/L, and 800 nmol/L, while maintaining other parameters constant. Finally, the reaction time was optimized by varying it among incubation of 5 min, 10 min, 15 min, 20 min, and 30 min, followed by the assessment of fluorescence signal intensity under the diverse time conditions.
Using the optimized reaction conditions obtained above, the sensitivity of the RPA-CRISPR/Cas12a-FRB assay was further evaluated. DNA was serially diluted 10-fold to concentrations of 1 ng/μL, 0.1 ng/μL, 10 pg/μL, 1 pg/μL, 0.1 pg/μL, and 0.01 pg/μL. Each sample was tested in triplicate with sterile water as the negative control.
2.6. CRISPR/Cas12a-PLFS Visual Detection
The RPA-CRISPR/Cas12a-PLFS assay system contains 2 μL RPA product, 13.2 μL DEPC-H
2O, 2 μL NEBuffer (10×), 0.4 μL Cas12a (5 μmol/L, New England Biolabs, Ipswich, MA, USA), 0.4 μL St-crRNA (0.01 mmol/L), 2 μL LF-DNA reporter (5 μmol/L). After incubating under 37 °C for 20 min to perform CRISPR/Cas12a cleavage assay, 80 μL DEPC-H
2O was added to the reaction tube and mixed well. Then, the lateral flow strip (Suzhou Gendx Biotech Co., Ltd., Suzhou, China) was inserted into the tube and incubated at room temperature for 7 min. A positive result is indicated by the presence of a red band at the test line or at both the test line and control line on the strip. Conversely, a negative result is indicated by the absence of a red band at the test line but the presence of a red band at the control line (
Figure 2d).
Subsequently, to optimize the reaction conditions of the CRISPR/Cas12a-PLFS system, the concentration of the reporter in the 100 μL reaction system was fine-tuned, with the LF-DNA concentrations set at 50 nmol/L, 100 nmol/L, 200 nmol/L, 400 nmol/L, and 800 nmol/L, while maintaining other parameters constant. Subsequently, the reaction time was optimized by varying it among incubation of 5 min, 10 min, 15 min, 20 min, and 30 min.
Using the optimized reaction conditions obtained above, the sensitivity of the RPA-CRISPR/Cas12a-PLFS assay was further evaluated. DNA was serially diluted 10-fold to concentrations of 1 ng/μL, 0.1 ng/μL, 10 pg/μL, 1 pg/μL, 0.1 pg/μL, and 0.01 pg/μL. Each sample was tested in triplicate with sterile water as the negative control.
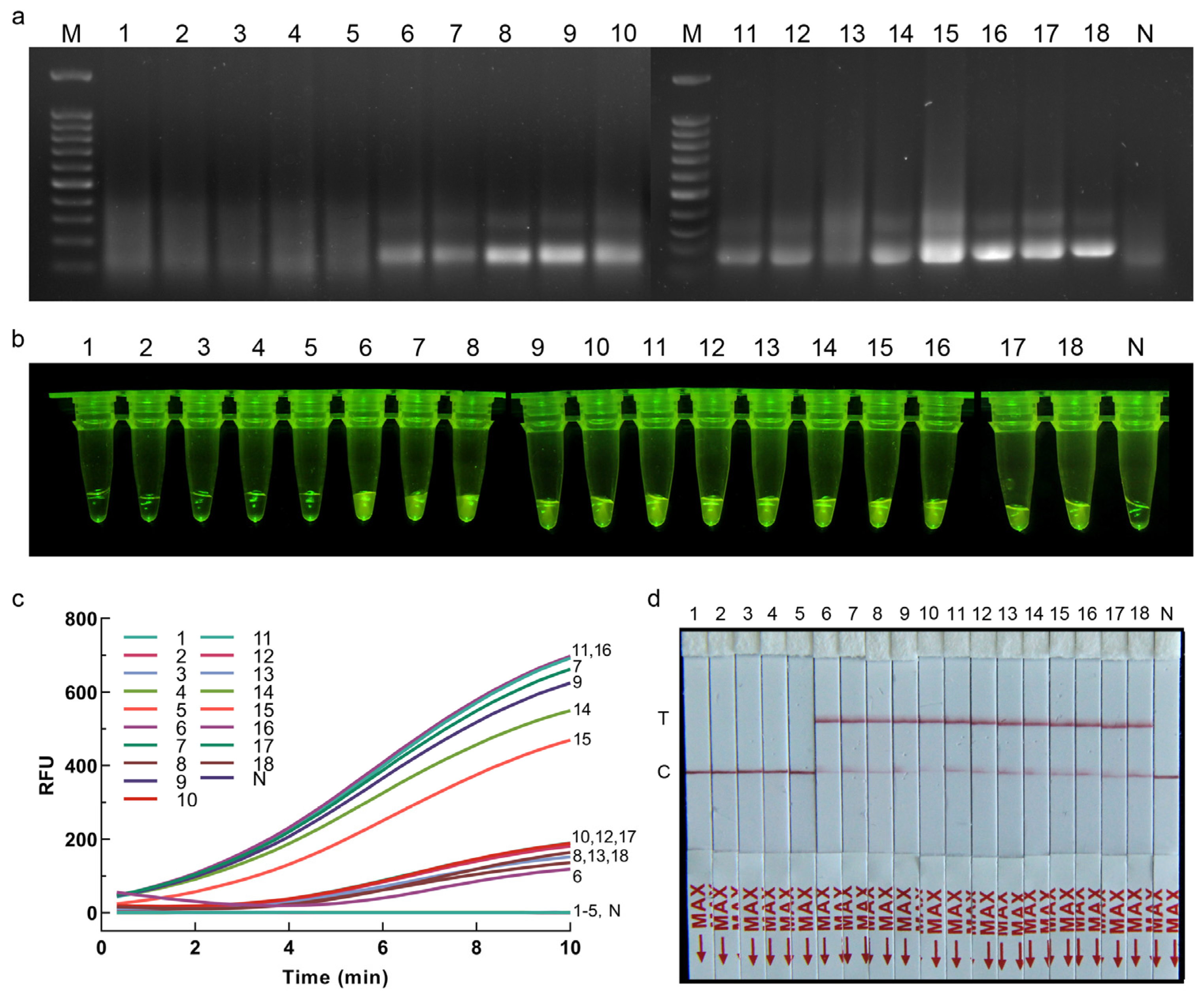
2.7. Application of RPA-CRISPR/Cas12a Assay for Plant Samples
Application of the RPA-CRISPR/Cas12a assay for plant samples was assessed using inoculated and healthy onions. To obtain the plant samples, the strain for inoculation and the onion samples to be inoculated should be prepared at the same time. Seventeen purchased healthy onions were hydroponically grown, and the pathogen inoculation test was carried out after the growth of fibrous roots. Strain CBS 335.29 was cultured on PDA medium for 15 d. Agar blocks (5 mm in diameter) carrying mycelia were then cut from PDA plates and placed on the fibrous roots of twelve onions that were stabbed with a sterile inoculation needle. Five onions inoculated with sterile agar blocks were used as control. The onions were then placed in 9 cm Petri dishes with four layers of moisturizing gauze at the bottom and incubated at 25 °C. After 7 d of inoculation, one fibrous root sample from each onion was cut from the inoculated and control groups. The DNA of each root sample was extracted separately as the detection template using a modified CTAB protocol [
19], and the genomic DNA of strain CBS 335.29 was used as the positive control, and sterile water instead of template was used as the negative control for the RPA-CRISPR/Cas12a assay in order to evaluate the effectiveness of the detection methods. The optimized reaction conditions of RPA-CRISPR/Cas12a-FRB and RPA-CRISPR/Cas12a-PLFS were used for the detection of plant samples.
4. Discussion
The increasing globalization of trade represents a significant threat due to the spread of invasive alien phytopathogens through various pathways, including cargo, passengers, conveyances, postal services, wooden packaging and containers [
20].
Setophoma terrestris, a common soil resident, is responsible for causing pink root rot in multiple crops, including onion, muskmelon, watermelon, maize, squash, and canola plants [
3,
21,
22,
23,
24]. The intensification of global trade has increased the risk of introducing
S. terrestris into new regions, emphasizing the necessity for the efficient identification and management of this plant pathogen in order to safeguard agricultural interests. While Yoshida [
25] established a SYBR Green qPCR assay for this pathogen, it is important to note that SYBR Green can bind non-specifically to the small groove of double-stranded DNA, leading to challenges in differentiating primer dimers or non-specific amplification products, and susceptibility to false positives and fluorescent signals being detected, often necessitating a combination with a melting curve analysis to exclude false positives. Likewise, in an effort to enhance the accuracy of the assay, Li et al. [
11] established a TaqMan MGB-based qPCR. However, both detection assays have high equipment requirements and face challenges in meeting the demand for on-site rapid testing, presenting significant limitations. In contrast, the RPA operates within a stable temperature range of 37 to 42 °C, making it accessible through various means such as warm water, an incubator, room temperature, or even body heat. The compatibility of RPA with CRISPR-Cas12a detection is promising, given their congruent temperature requirements.
This study introduced two rapid and precise visual detection methods for S. terrestris, namely RPA-CRISPR/Cas12a-FRB and RPA-CRISPR/Cas12a-PLFS. The RPA-CRISPR/Cas12a-FRB approach can accurately detect S. terrestris within a 30 min timeframe, boasting a sensitivity of up to 0.01 pg/μL. Contrasted with the RPA-CRISPR/Cas12a-FRB system, the RPA-CRISPR/Cas12a-PLFS method can deliver precise results within 30 min, without the necessity of additional blue light equipment, featuring a sensitivity of 0.01 pg/μL. When considering quick inspections at customs, a rapid DNA extraction method can be used as an alternative before applying RPA-CRISPR/Cas12a assays, i.e., placing the hyphae in a centrifuge tube containing 50 μL of sterile ddH2O, heating it at 95 °C for 10 min, and then centrifuging to obtain the supernatant. If the rapid DNA extraction method mentioned in this study is employed, the entire detection process will not exceed 40 min, including DNA extraction, RPA reaction, Cas12a cleavage, and result readout.
In the RPA-CRISPR/Cas12a detection assay, we first evaluated the optimal RPA reaction time and temperature based on the gel electrophoresis band brightness of the RPA reaction product. This is mainly because the band brightness is related to the concentration of the amplification product, and the concentration directly affects the sensitivity of the CRISPR/Cas12a assay. In this study, we also tried to reduce the overall RPA-CRISPR/Cas12a detection process by shortening the RPA reaction time. However, when the RPA reaction time was shortened to 10 min, the sensitivity of the subsequent CRISPR/Cas12a visual detection method could only reach 0.1 ng. Therefore, the RPA reaction time of 20 min was chosen for this study in order to balance the detection effect and duration simultaneously.
When compared to real-time PCR, the developed RPA-CRISPR/Cas12a system offers simpler procedures, milder conditions, more adaptable signal readouts, and notably higher sensitivity, as the established minimal detection limit of the real-time PCR method for
S. terrestris is 1 pg/uL [
11]. Therefore, the visual detection approaches outlined in this study enable rapid and accurate screening for
S. terrestris at ports and agricultural sites, mitigating the spread through the import, export, and transport of seeds and other reproductive materials. However, in comparison to real-time PCR, the RPA-CRISPR/Cas12a-FRB and RPA-CRISPR/Cas12a-PLFS methods present a disadvantage in terms of cost, with a price that is approximately 3–5 times higher than that of real-time PCR detection methods. This is primarily due to the relatively higher cost of enzymes. Nevertheless, it is anticipated that with the progression of technology, the expense of enzymes will be diminished in the future, rendering this method more convenient than real-time PCR.
The ITS sequence of Setophoma is distinctly different from that of other genera, ensuring specificity in primer design for this study. Therefore, strains from other genera were not used for verification during the experimental verification stage. Additionally, the total DNA of onion tissue also includes DNA from other microbial groups. The fact that healthy onion tissue did not yield positive results in the RPA-CRISPR/Cas12a assays further confirms the reliability of this study.
Nonetheless, we acknowledge that this study has certain limitations. One of the important issues is the restricted number and diversity of S. terrestris samples included in this study for testing primers, largely due to challenges in accessing materials for studying imported quarantine fungi in China. Despite this limitation, our sequence similarity comparisons and phylogenetic analyses indicate distinct ITS region sequence differences between S. terrestris and its close relatives. Furthermore, ITS sequences from strains originating from different countries show relative conservation. Therefore, we are confident that the primers developed in this study are specific and expect to further validate the validity and sensitivity of the RPA-CRISPR/Cas12a assay through international collaboration. In addition, as S. terrestris is a quarantine fungus for entry into China, no diseased plants infected by this species have been found in the field. Consequently, it is not currently possible to conduct tests on real field-infested material. However, the tests conducted using artificially inoculated onion tissues provide evidence that the detection method developed in this study is feasible.





 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}