
Contemporary Diagnosis and Management of Hypertrophic Cardiomyopathy: The Role of Echocardiography and Multimodality Imaging

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Diagnosis and Variation of Hypertrophy
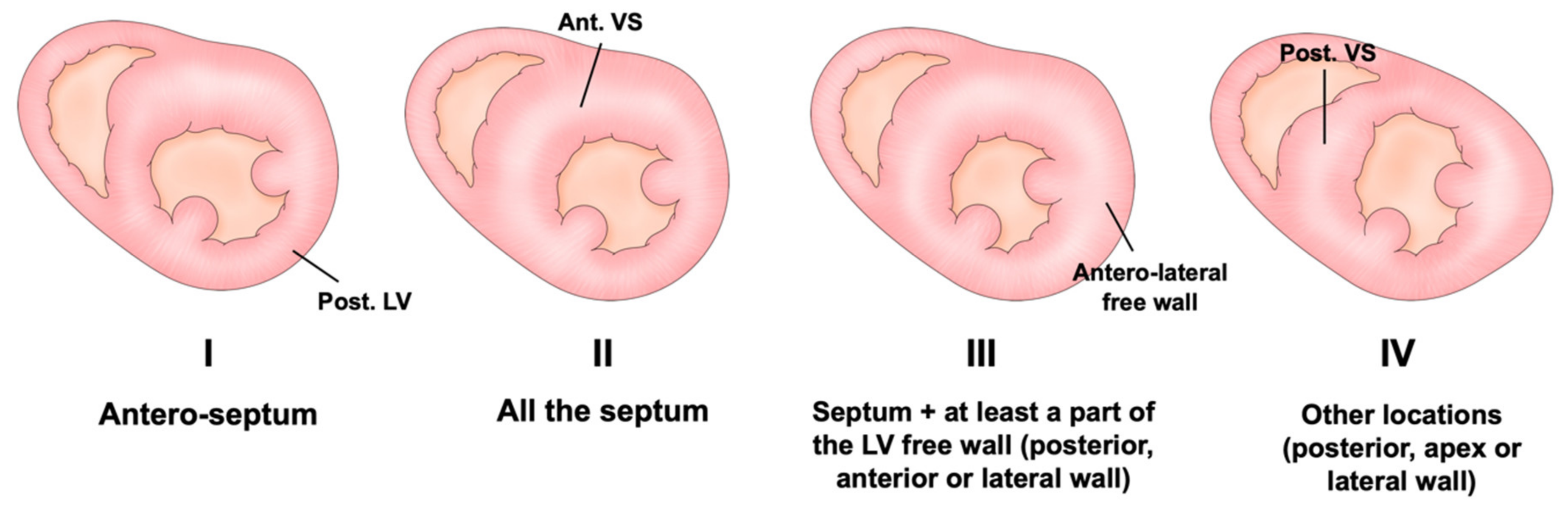
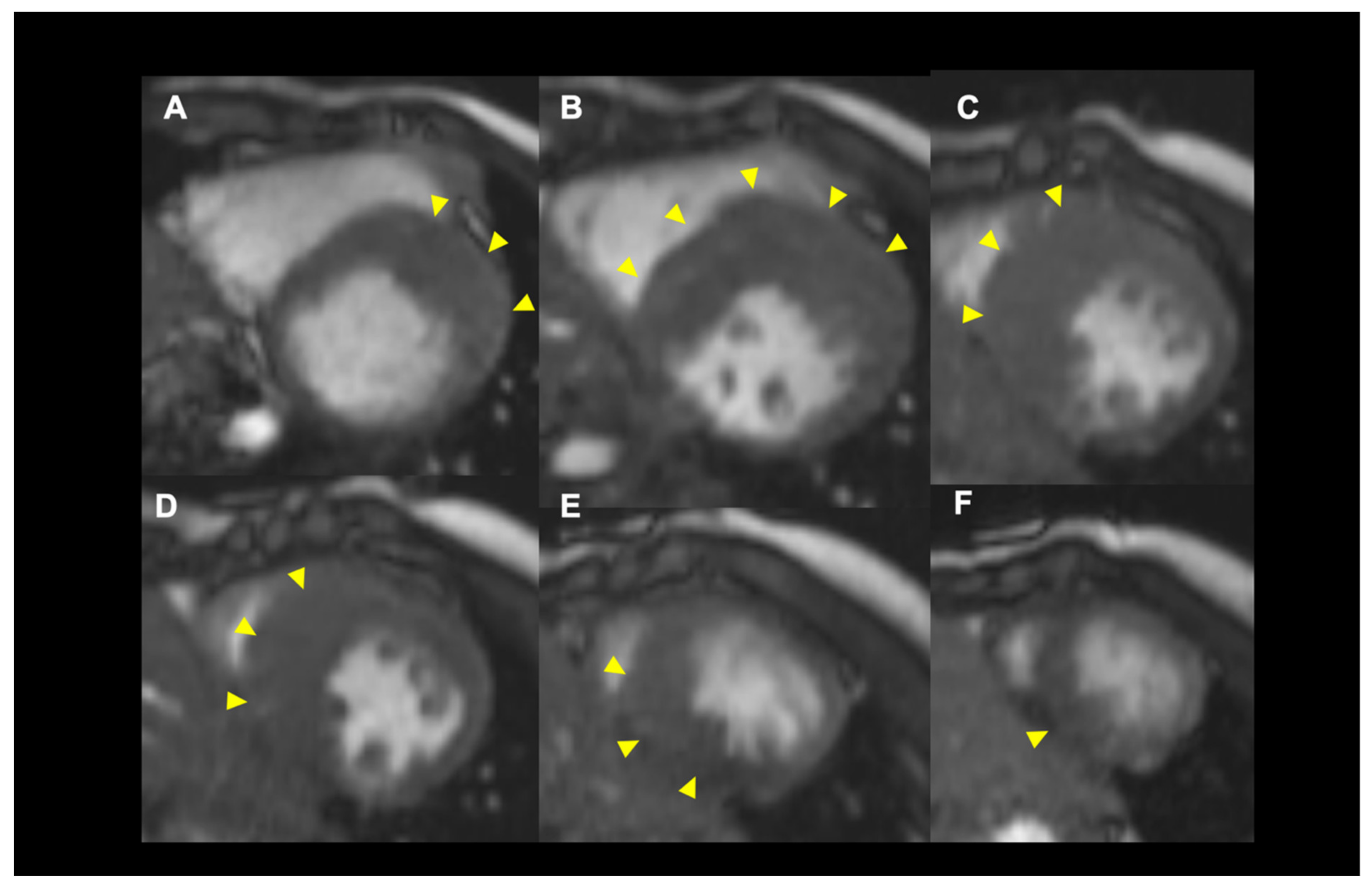
Assessment of LV Wall Hypertrophy
3. Various Types of Left Ventricular Structural Abnormality
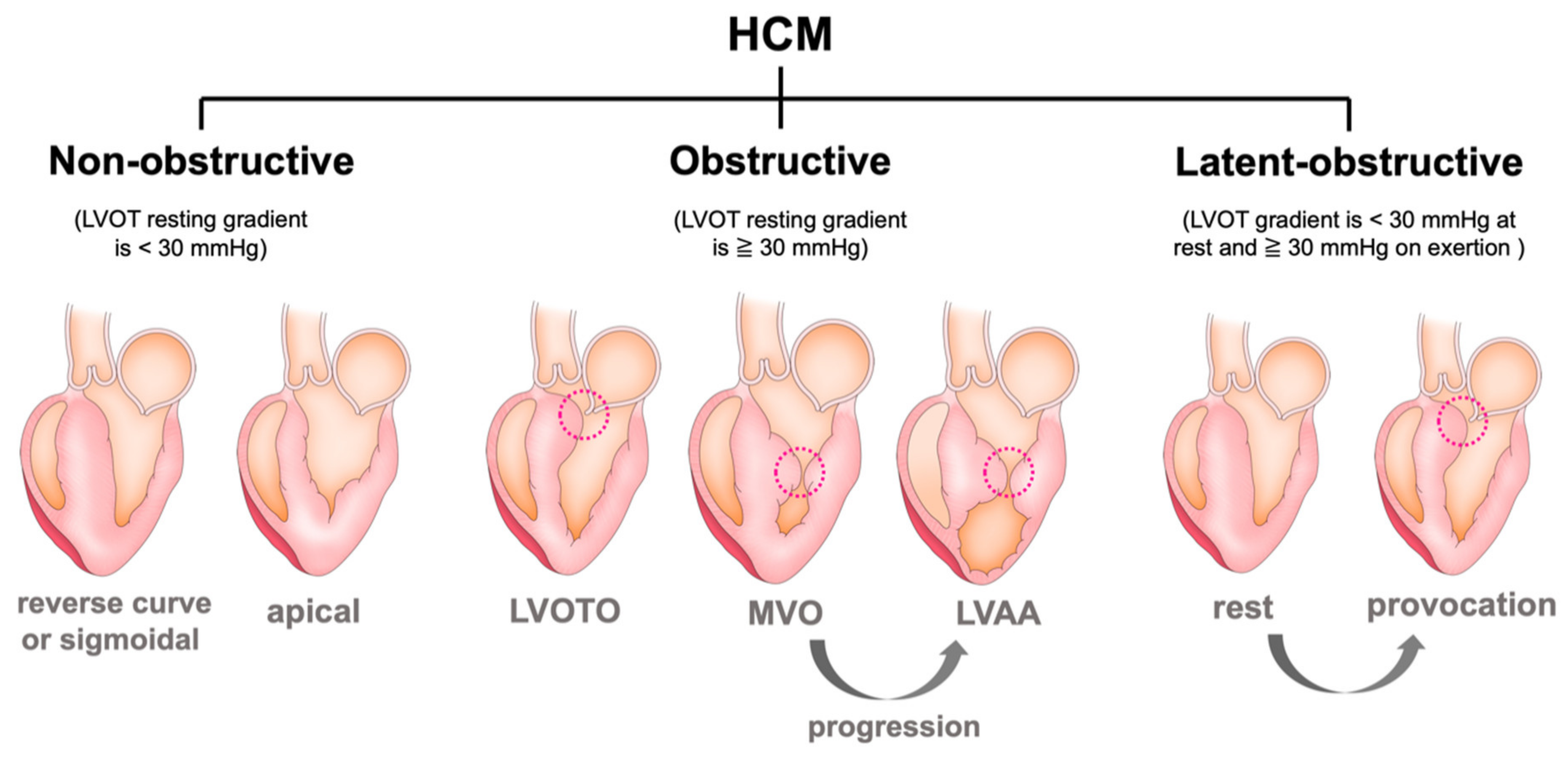
3.1. Left Ventricular Outflow Obstruction (LVOTO)
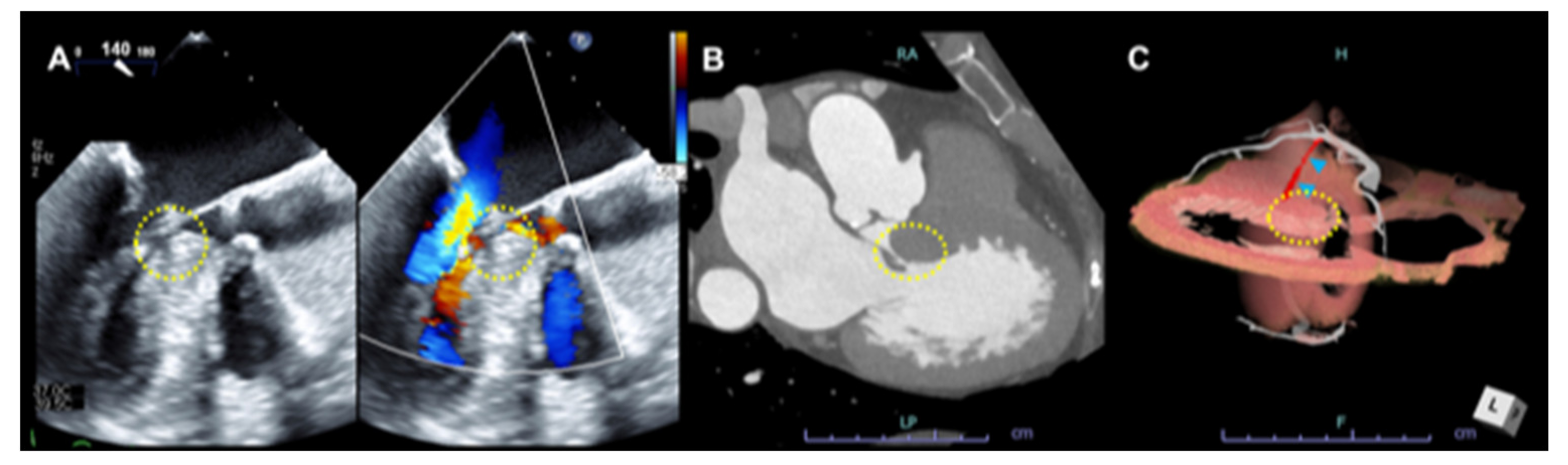
3.2. Systolic Anterior Motion (SAM) of the Mitral Valve and Anomaly in the Mitral Valve Apparatus
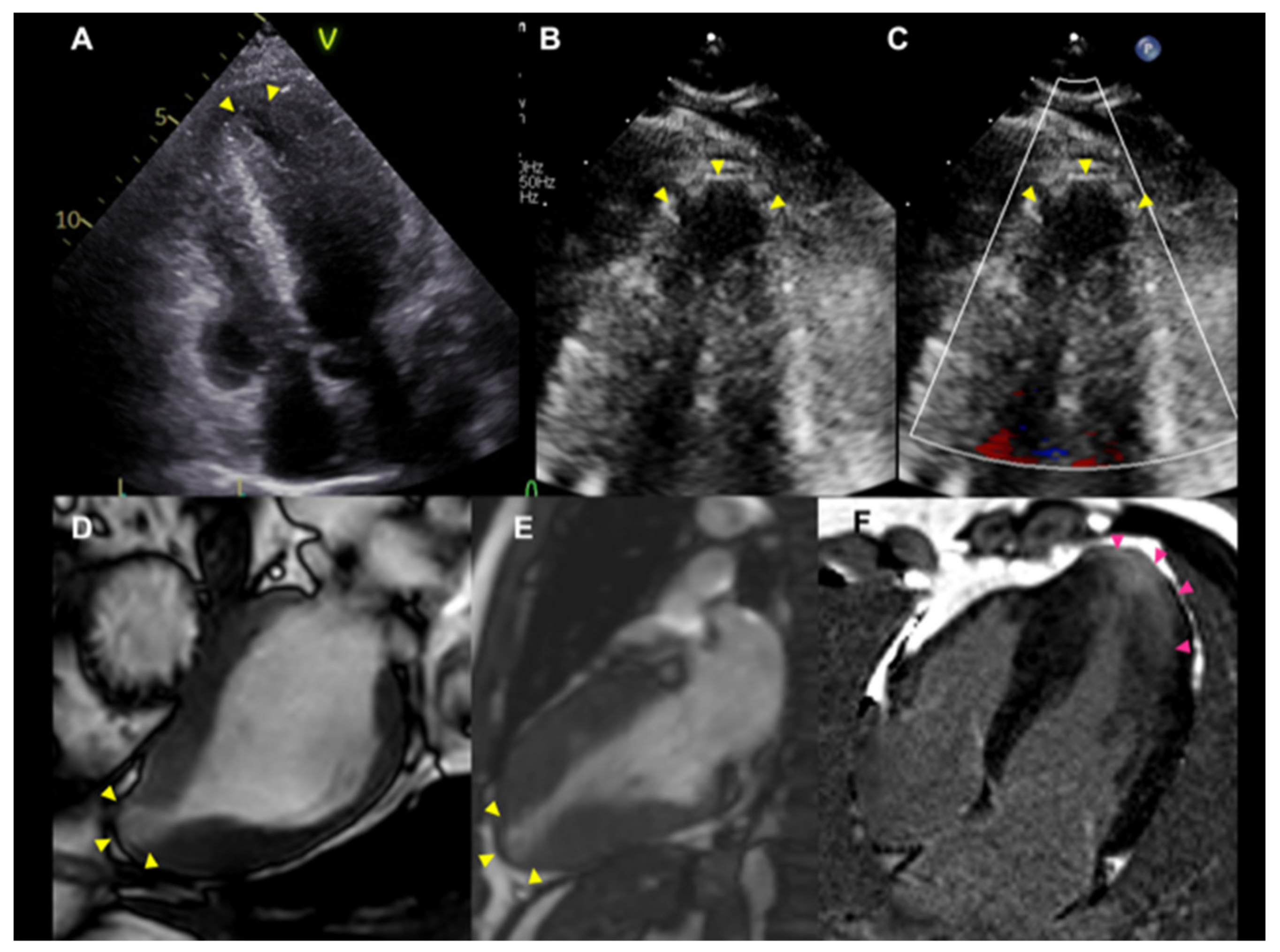
3.3. Mid-Ventricular Obstruction (MVO) and Left Ventricular Apical Aneurysm (LVAA)
3.4. Reduced LV Function (Dilated Phase/End-Stage/Advanced Stage)
4. Risk of Sudden Cardiac Death
4.1. Risk Scores
4.2. Role of Cardiac MRI for SCD Risk Stratification
4.3. Novel Echocardiographic Technique
5. Role of Pharmacological Therapy
New Drugs
6. Role of Genetic Testing
7. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults. Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef]
- Hada, Y.; Sakamoto, T.; Amano, K.; Yamaguchi, T.; Takenaka, K.; Takahashi, H.; Takikawa, R.; Hasegawa, I.; Takahashi, T.; Suzuki, J. Prevalence of Hypertrophic Cardiomyopathy in a Population of Adult Japanese Workers as Detected by Echocardiographic Screening. Am. J. Cardiol. 1987, 59, 183–184. [Google Scholar] [CrossRef]
- Maron, M.S.; Hellawell, J.L.; Lucove, J.C.; Farzaneh-Far, R.; Olivotto, I. Occurrence of Clinically Diagnosed Hypertrophic Cardiomyopathy in the United States. Am. J. Cardiol. 2016, 117, 1651–1654. [Google Scholar] [CrossRef]
- Maron, B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. G, American Association for Thoracic S, American Society of E, American Society of Nuclear C, Heart Failure Society of A, Heart Rhythm S. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular A, Interventions and Society of Thoracic S. Circulation 2011, 124, e783–e831. [Google Scholar]
- Spirito, P.; Autore, C.; Formisano, F.; Assenza, G.E.; Biagini, E.; Haas, T.S.; Bongioanni, S.; Semsarian, C.; Devoto, E.; Musumeci, B.; et al. Risk of Sudden Death and Outcome in Patients with Hypertrophic Cardiomyopathy with Benign Presentation and Without Risk Factors. Am. J. Cardiol. 2014, 113, 1550–1555. [Google Scholar] [CrossRef]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Haas, T.S.; Chan, R.H.; Udelson, J.E.; Garberich, R.F.; Lesser, J.R.; Appelbaum, E.; Manning, W.J.; et al. Risk Stratification and Outcome of Patients with Hypertrophic Cardiomyopathy >=60 Years of Age. Circulation 2013, 127, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Rowin, E.J.; Hausvater, A.; Link, M.S.; Abt, P.; Gionfriddo, W.; Wang, W.; Rastegar, H.; Estes, N.A.M.; Maron, M.S.; Maron, B.J. Clinical Profile and Consequences of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation 2017, 136, 2420–2436. [Google Scholar] [CrossRef] [PubMed]
- Afonso, L.C.; Bernal, J.; Bax, J.J.; Abraham, T.P. Echocardiography in Hypertrophic Cardiomyopathy: The Role of Conventional and Emerging Technologies. JACC Cardiovasc. Imaging 2008, 1, 787–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardim, N.; Galderisi, M.; Edvardsen, T.; Plein, S.; Popescu, B.A.; D’Andrea, A.; Bruder, O.; Cosyns, B.; Davin, L.; Donal, E.; et al. Role of Multimodality Cardiac Imaging in the Management of Patients with Hypertrophic Cardiomyopathy: An Expert Consensus of the European Association of Cardiovascular Imaging Endorsed by the Saudi Heart Association. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.K.; Frenneaux, M.P.; Steeds, R.P. Echocardiography in Hypertrophic Cardiomyopathy Diagnosis, Prognosis, and Role in Management. Eur. J. Echocardiogr. 2009, 10, iii9–iii14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.J.; Gottdiener, J.S.; Epstein, S.E. Patterns and Significance of Distribution of Left Ventricular Hypertrophy in Hypertrophic Cardiomyopathy. A Wide Angle, Two Dimensional Echocardiographic Study of 125 Patients. Am. J. Cardiol. 1981, 48, 418–428. [Google Scholar] [CrossRef]
- Syed, I.S.; Ommen, S.R.; Breen, J.F.; Tajik, A.J. Hypertrophic Cardiomyopathy: Identification of Morphological Subtypes by Echocardiography and Cardiac Magnetic Resonance Imaging. JACC Cardiovasc. Imaging 2008, 1, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Helmy, S.M.; Maauof, G.F.; Shaaban, A.A.; Elmaghraby, A.M.; Anilkumar, S.; Shawky, A.H.; Hajar, R. Hypertrophic Cardiomyopathy: Prevalence, Hypertrophy Patterns, and Their Clinical and ECG Findings in a Hospital at Qatar. Heart Views 2011, 12, 143–149. [Google Scholar] [CrossRef]
- Swoboda, P.P.; McDiarmid, A.K.; Erhayiem, B.; Broadbent, D.A.; Dobson, L.E.; Garg, P.; Ferguson, C.; Page, S.P.; Greenwood, J.P.; Plein, S. Assessing Myocardial Extracellular Volume by T1 Mapping to Distinguish Hypertrophic Cardiomyopathy from Athlete’s Heart. J. Am. Coll. Cardiol. 2016, 67, 2189–2190. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of Left Ventricular Outflow Tract Obstruction on Clinical Outcome in Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef]
- Maron, M.S.; Rowin, E.J.; Olivotto, I.; Casey, S.A.; Arretini, A.; Tomberli, B.; Garberich, R.F.; Link, M.S.; Chan, R.H.M.; Lesser, J.R.; et al. Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 1399–1409. [Google Scholar] [CrossRef]
- Olivotto, I.; Maron, M.S.; Adabag, A.S.; Casey, S.A.; Vargiu, D.; Link, M.S.; Udelson, J.E.; Cecchi, F.; Maron, B.J. Gender-Related Differences in the Clinical Presentation and Outcome of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 46, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowin, E.J.; Maron, B.J.; Olivotto, I.; Maron, M.S. Role of Exercise Testing in Hypertrophic Cardiomyopathy. JACC Cardiovasc. Imaging 2017, 10, 1374–1386. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.A.; Vlahakes, G.J.; Lefebvre, X.; Guerrero, J.L.; Cape, E.G.; Yoganathan, A.P.; Weyman, A.E. Papillary Muscle Displacement Causes Systolic Anterior Motion of the Mitral Valve. Experimental Validation and Insights into the Mechanism of Subaortic Obstruction. Circulation 1995, 91, 1189–1195. [Google Scholar] [CrossRef]
- Veselka, J.; Krejci, J.; Tomasov, P.; Zemanek, D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: A comparison with general population. Eur. Heart J. 2014, 35, 2040–2045. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.; Rao, C.; Ashrafian, H.; Chaudhry, U.; Darzi, A.; Athanasiou, T. Modern Management of Systolic Anterior Motion of the Mitral Valve. Eur. J. Cardiothorac. Surg. 2012, 41, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.H.; Omran, A.S.; Wigle, E.D.; Williams, W.G.; Siu, S.C.; Rakowski, H. Mitral Regurgitation in Hypertrophic Obstructive Cardiomyopathy: Relationship to Obstruction and Relief with Myectomy. J. Am. Coll. Cardiol. 2000, 36, 2219–2225. [Google Scholar] [CrossRef] [Green Version]
- Kizilbash, A.M.; Heinle, S.K.; Grayburn, P.A. Spontaneous Variability of Left Ventricular Outflow Tract Gradient in Hypertrophic Obstructive Cardiomyopathy. Circulation 1998, 97, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Rowin, E.J.; Maron, B.J.; Haas, T.S.; Garberich, R.F.; Wang, W.; Link, M.S.; Maron, M.S. Hypertrophic Cardiomyopathy with Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J. Am. Coll. Cardiol. 2017, 69, 761–773. [Google Scholar] [CrossRef]
- Autore, C.; Musumeci, M.B. The Natural History of Hypertrophic Cardiomyopathy. Eur. Heart J. Suppl. 2020, 22, L11–L14. [Google Scholar] [CrossRef]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Maron, M.S. How Hypertrophic Cardiomyopathy Became a Contemporary Treatable Genetic Disease with Low Mortality: Shaped by 50 Years of Clinical Research and Practice. JAMA Cardiol. 2016, 1, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Haland, T.F.; Hasselberg, N.E.; Almaas, V.M.; Dejgaard, L.A.; Saberniak, J.; Leren, I.S.; Berge, K.E.; Haugaa, K.H.; Edvardsen, T. The Systolic Paradox in Hypertrophic Cardiomyopathy. Open Heart 2017, 4, e000571. [Google Scholar] [CrossRef] [PubMed]
- Tower-Rader, A.; Mohananey, D.; To, A.; Lever, H.M.; Popovic, Z.B.; Desai, M.Y. Prognostic Value of Global Longitudinal Strain in Hypertrophic Cardiomyopathy: A Systematic Review of Existing Literature. JACC Cardiovasc. Imaging 2019, 12, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Cecchi, F.; Poggesi, C.; Yacoub, M.H. Patterns of Disease Progression in Hypertrophic Cardiomyopathy: An Individualized Approach to Clinical Staging. Circ. Heart Fail. 2012, 5, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, M.B.; Russo, D.; Limite, L.R.; Canepa, M.; Tini, G.; Casenghi, M.; Francia, P.; Adduci, C.; Pagannone, E.; Magrì, D.; et al. Long-Term Left Ventricular Remodeling of Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 1924–1931. [Google Scholar] [CrossRef]
- Maron, B.J.; Spirito, P. Implications of Left Ventricular Remodeling in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 1998, 81, 1339–1344. [Google Scholar] [CrossRef]
- Harris, K.M.; Spirito, P.; Maron, M.S.; Zenovich, A.G.; Formisano, F.; Lesser, J.R.; Mackey-Bojack, S.; Manning, W.J.; Udelson, J.E.; Maron, B.J. Prevalence, Clinical Profile, and Significance of Left Ventricular Remodeling in the End-Stage Phase of Hypertrophic Cardiomyopathy. Circulation 2006, 114, 216–225. [Google Scholar] [CrossRef]
- Kubo, T.; Gimeno, J.R.; Bahl, A.; Steffensen, U.; Steffensen, M.; Osman, E.; Thaman, R.; Mogensen, J.; Elliott, P.M.; Doi, Y.; et al. Prevalence, Clinical Significance, and Genetic Basis of Hypertrophic Cardiomyopathy with Restrictive Phenotype. J. Am. Coll. Cardiol. 2007, 49, 2419–2426. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Pencina, M.J.; Assenza, G.E.; Haas, T.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; et al. Prognostic Value of Quantitative Contrast-Enhanced Cardiovascular Magnetic Resonance for the Evaluation of Sudden Death Risk in Patients with Hypertrophic Cardiomyopathy. Circulation 2014, 130, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.M.; Gimeno, J.R.; Thaman, R.; Shah, J.; Ward, D.; Dickie, S.; Tome Esteban, M.T.; McKenna, W.J. Historical Trends in Reported Survival Rates in Patients with Hypertrophic Cardiomyopathy. Heart 2006, 92, 785–791. [Google Scholar] [CrossRef]
- Ammirati, E.; Contri, R.; Coppini, R.; Cecchi, F.; Frigerio, M.; Olivotto, I. Pharmacological Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives. Eur. J. Heart Fail. 2016, 18, 1106–1118. [Google Scholar] [CrossRef]
- Elliott, P.M.; Poloniecki, J.; Dickie, S.; Sharma, S.; Monserrat, L.; Varnava, A.; Mahon, N.G.; McKenna, W.J. Sudden Death in Hypertrophic Cardiomyopathy: Identification of High Risk Patients. J. Am. Coll. Cardiol. 2000, 36, 2212–2218. [Google Scholar] [CrossRef]
- Weissler-Snir, A.; Allan, K.; Cunningham, K.; Connelly, K.A.; Lee, D.S.; Spears, D.A.; Rakowski, H.; Dorian, P. Hypertrophic Cardiomyopathy-Related Sudden Cardiac Death in Young People in Ontario. Circulation 2019, 140, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. IA Novel Clinical Risk Prediction Model for Sudden Cardiac Death in Hypertrophic Cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Maron, B.J.; Casey, S.A.; Chan, R.H.; Garberich, R.F.; Rowin, E.J.; Maron, M.S. Independent Assessment of the European Society of Cardiology Sudden Death Risk Model for Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Fraiche, A.; Wang, A. Hypertrophic Cardiomyopathy: New Evidence since the 2011 American Cardiology of Cardiology Foundation and American Heart Association Guideline. Curr. Cardiol. Rep. 2016, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Okada, A.; Nishimura, K.; Hamatani, Y.; Amano, M.; Takahama, H.; Amaki, M.; Hasegawa, T.; Kanzaki, H.; Kusano, K.; et al. Validation of the 2014 European Society of Cardiology Sudden Cardiac Death Risk Prediction Model Among Various Phenotypes in Japanese Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2018, 122, 1939–1946. [Google Scholar] [CrossRef]
- O’Mahony, C.; Akhtar, M.M.; Anastasiou, Z.; Guttmann, O.P.; Vriesendorp, P.A.; Michels, M.; Magrì, D.; Autore, C.; Fernández, A.; Ochoa, J.P.; et al. Effectiveness of the 2014 European Society of Cardiology Guideline on Sudden Cardiac Death in Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. Heart 2019, 105, 623–631. [Google Scholar] [CrossRef]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of Left Ventricular Hypertrophy and Risk of Sudden Death in Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef]
- Popescu, B.A.; Rosca, M.; Schwammenthal, E. Dynamic Obstruction in Hypertrophic Cardiomyopathy. Curr. Opin. Cardiol. 2015, 30, 468–474. [Google Scholar] [CrossRef]
- Wang, W.; Lian, Z.; Rowin, E.J.; Maron, B.J.; Maron, M.S.; Link, M.S. Prognostic Implications of Nonsustained Ventricular Tachycardia in High-Risk Patients with Hypertrophic Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2017, 10, e004604. [Google Scholar] [CrossRef]
- Chan, R.H.; Maron, B.J.; Olivotto, I.; Assenza, G.E.; Haas, T.S.; Lesser, J.R.; Gruner, C.; Crean, A.M.; Rakowski, H.; Rowin, E.; et al. Significance of Late Gadolinium Enhancement at Right Ventricular Attachment to Ventricular Septum in Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2015, 116, 436–441. [Google Scholar] [CrossRef]
- Avanesov, M.; Münch, J.; Weinrich, J.; Well, L.; Säring, D.; Stehning, C.; Tahir, E.; Bohnen, S.; Radunski, U.K.; Muellerleile, K.; et al. Prediction of the Estimated 5-Year Risk of Sudden Cardiac Death and Syncope or Non-Sustained Ventricular Tachycardia in Patients with Hypertrophic Cardiomyopathy Using Late Gadolinium Enhancement and Extracellular Volume CMR. Eur. Radiol. 2017, 27, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Hauser, T.H.; Dubrow, E.; Horst, T.A.; Kissinger, K.V.; Udelson, J.E.; Manning, W.J. Right Ventricular Involvement in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2007, 100, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Bierig, S.M.; Budoff, M.J.; Desai, M.; Dilsizian, V.; Eidem, B.; Goldstein, S.A.; Hung, J.; Maron, M.S.; Ommen, S.R.; et al. American Society of Echocardiography Clinical Recommendations for Multimodality Cardiovascular Imaging of Patients with Hypertrophic Cardiomyopathy: Endorsed by the American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. J. Am. Soc. Echocardiogr. 2011, 24, 473–498. [Google Scholar] [CrossRef]
- Petersen, S.E.; Jerosch-Herold, M.; Hudsmith, L.E.; Robson, M.D.; Francis, J.M.; Doll, H.A.; Selvanayagam, J.B.; Neubauer, S.; Watkins, H. Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: New insights from multiparametric magnetic resonance imaging. Circulation 2007, 115, 2418–2425. [Google Scholar] [CrossRef] [Green Version]
- Loong, C.Y.; Reyes, E.; Underwood, S.R. Significant inducible perfusion abnormality in an asymptomatic patient with hypertrophic cardiomyopathy demonstrated by radionuclide myocardial perfusion imaging. Heart 2003, 89, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaoka, H.; Kubo, T.; Okawa, M.; Takenaka, N.; Sakamoto, C.; Baba, Y.; Hayashi, K.; Yamasaki, N.; Matsumura, Y.; Doi, Y.L. Tissue Doppler Imaging and Plasma BNP Levels to Assess the Prognosis in Patients with Hypertrophic Cardiomyopathy. J. Am. Soc. Echocardiogr. 2011, 24, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Take, Y.; Morita, H.; Toh, N.; Nishii, N.; Nagase, S.; Nakamura, K.; Kusano, K.F.; Ohe, T.; Ito, H. Identification of High-Risk Syncope Related to Ventricular Fibrillation in Patients with Brugada Syndrome. Heart Rhythm 2012, 9, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Haland, T.F.; Almaas, V.M.; Hasselberg, N.E.; Saberniak, J.; Leren, I.S.; Hopp, E.; Edvardsen, T.; Haugaa, K.H. Strain Echocardiography Is Related to Fibrosis and Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvari, S.I.; Haugaa, K.H.; Anfinsen, O.G.; Leren, T.P.; Smiseth, O.A.; Kongsgaard, E.; Amlie, J.P.; Edvardsen, T. Right Ventricular Mechanical Dispersion Is Related to Malignant Arrhythmias: A Study of Patients with Arrhythmogenic Right Ventricular Cardiomyopathy and Subclinical Right Ventricular Dysfunction. Eur. Heart J. 2011, 32, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Haugaa, K.H.; Smedsrud, M.K.; Steen, T.; Kongsgaard, E.; Loennechen, J.P.; Skjaerpe, T.; Voigt, J.U.; Willems, R.; Smith, G.; Smiseth, O.A.; et al. Mechanical Dispersion Assessed by Myocardial Strain in Patients After Myocardial Infarction for Risk Prediction of Ventricular Arrhythmia. JACC Cardiovasc. Imaging 2010, 3, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugaa, K.H.; Goebel, B.; Dahlslett, T.; Meyer, K.; Jung, C.; Lauten, A.; Figulla, H.R.; Poerner, T.C.; Edvardsen, T. Risk Assessment of Ventricular Arrhythmias in Patients with Nonischemic Dilated Cardiomyopathy by Strain Echocardiography. J. Am. Soc. Echocardiogr. 2012, 25, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; Olivotto, I.; Maron, M.S.; Ferrantini, C.; Coppini, R.; Grifoni, C.; Baldini, K.; Sgalambro, A.; Cecchi, F.; Maron, B.J. Beta Blockers for Prevention of Exercise-Induced Left Ventricular Outflow Tract Obstruction in Patients with Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2012, 110, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Ostman-Smith, I.; Wettrell, G.; Riesenfeld, T. A Cohort Study of Childhood Hypertrophic Cardiomyopathy: Improved Survival Following High-Dose Beta-Adrenoceptor Antagonist Treatment. J. Am. Coll. Cardiol. 1999, 34, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Tendera, M.; Wycisk, A.; Schneeweiss, A.; Poloński, L.; Wodniecki, J. Effect of Sotalol on Arrhythmias and Exercise Tolerance in Patients with Hypertrophic Cardiomyopathy. Cardiology 1993, 82, 335–342. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Barac, I.; McKenna, W.J.; Elliott, P.M.; Dickie, S.; Chojnowska, L.; Casey, S.; Maron, B.J. Multicenter Study of the Efficacy and Safety of Disopyramide in Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.; Fourey, D.; Weissler-Snir, A.; Hindieh, W.; Chan, R.H.; Gollob, M.H.; Rakowski, H. Safety of Outpatient Initiation of Disopyramide for Obstructive Hypertrophic Cardiomyopathy Patients. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the Super Relaxed State of Human Beta-Cardiac Myosin and the Mode of Action of Mavacamten from Myosin Molecules to Muscle Fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [Green Version]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for Treatment of Symptomatic Obstructive Hypertrophic Cardiomyopathy (Explorer-HCM): A Randomised, Double-Blind, Placebo-Controlled, phase 3 Trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Saberi, S.; Cardim, N.; Yamani, M.; Schulz-Menger, J.; Li, W.; Florea, V.; Sehnert, A.J.; Kwong, R.Y.; Jerosch-Herold, M.; Masri, A.; et al. Mavacamten Favorably Impacts Cardiac Structure in Obstructive Hypertrophic Cardiomyopathy: Explorer-HCM Cardiac Magnetic Resonance Substudy Analysis. Circulation 2021, 143, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Fujino, N.; Anan, R.; Tei, C.; Kubo, T.; Doi, Y.; Kinugawa, S.; Tsutsui, H.; Kobayashi, S.; Yano, M.; et al. Sarcomere Gene Mutations Are Associated with Increased Cardiovascular Events in Left Ventricular Hypertrophy: Results from Multicenter Registration in Japan. JACC Heart Fail. 2013, 1, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gruner, C.; Chan, R.H.; Care, M.; Siminovitch, K.; Williams, L.; Woo, A.; Rakowski, H. Genotype-Positive Status in Patients with Hypertrophic Cardiomyopathy Is Associated with Higher Rates of Heart Failure Events. Circ. Cardiovasc. Genet. 2014, 7, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivotto, I.; Girolami, F.; Ackerman, M.J.; Nistri, S.; Bos, J.M.; Zachara, E.; Ommen, S.R.; Theis, J.L.; Vaubel, R.A.; Re, F.; et al. Myofilament Protein Gene Mutation Screening and Outcome of Patients with Hypertrophic Cardiomyopathy. Mayo Clin. Proc. 2008, 83, 630–638. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Harrigan, C.; Appelbaum, E.; Gibson, C.M.; Lesser, J.R.; Haas, T.S.; Udelson, J.E.; Manning, W.J.; Maron, B.J. Mitral Valve Abnormalities Identified by Cardiovascular Magnetic Resonance Represent a Primary Phenotypic Expression of Hypertrophic Cardiomyopathy. Circulation 2011, 124, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurizi, N.; Michels, M.; Rowin, E.J.; Semsarian, C.; Girolami, F.; Tomberli, B.; Cecchi, F.; Maron, M.S.; Olivotto, I.; Maron, B.J. Clinical Course and Significance of Hypertrophic Cardiomyopathy Without Left Ventricular Hypertrophy. Circulation 2019, 139, 830–833. [Google Scholar] [CrossRef]
- Bick, A.G.; Flannick, J.; Ito, K.; Cheng, S.; Vasan, R.S.; Parfenov, M.G.; Herman, D.S.; DePalma, S.R.; Gupta, N.; Gabriel, S.B.; et al. Burden of Rare Sarcomere Gene Variants in the Framingham and Jackson Heart Study Cohorts. Am. J. Hum. Genet. 2012, 91, 513–519. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitai, T.; Xanthopoulos, A.; Nakagawa, S.; Ishii, N.; Amano, M.; Triposkiadis, F.; Izumi, C. Contemporary Diagnosis and Management of Hypertrophic Cardiomyopathy: The Role of Echocardiography and Multimodality Imaging. J. Cardiovasc. Dev. Dis. 2022, 9, 169. https://doi.org/10.3390/jcdd9060169
Kitai T, Xanthopoulos A, Nakagawa S, Ishii N, Amano M, Triposkiadis F, Izumi C. Contemporary Diagnosis and Management of Hypertrophic Cardiomyopathy: The Role of Echocardiography and Multimodality Imaging. Journal of Cardiovascular Development and Disease. 2022; 9(6):169. https://doi.org/10.3390/jcdd9060169
Chicago/Turabian StyleKitai, Takeshi, Andrew Xanthopoulos, Shoko Nakagawa, Natsuko Ishii, Masashi Amano, Filippos Triposkiadis, and Chisato Izumi. 2022. "Contemporary Diagnosis and Management of Hypertrophic Cardiomyopathy: The Role of Echocardiography and Multimodality Imaging" Journal of Cardiovascular Development and Disease 9, no. 6: 169. https://doi.org/10.3390/jcdd9060169
APA StyleKitai, T., Xanthopoulos, A., Nakagawa, S., Ishii, N., Amano, M., Triposkiadis, F., & Izumi, C. (2022). Contemporary Diagnosis and Management of Hypertrophic Cardiomyopathy: The Role of Echocardiography and Multimodality Imaging. Journal of Cardiovascular Development and Disease, 9(6), 169. https://doi.org/10.3390/jcdd9060169