
Alternative Splicing of Pericentrin Contributes to Cell Cycle Control in Cardiomyocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Cell Culture of Postnatal Cardiomyocytes
2.2. Cell Culture of C2C12 Myoblasts and ARPE-19 Adult Retinal Pigment Epithelial Cells
2.3. SiRNA Knockdown
2.4. Analysis of DNA Incorporation
2.5. Reverse Transcriptase PCR (RT-PCR)
2.6. Plasmids
2.7. Plasmid Transfection
2.8. Immunofluorescence and Microscopy
2.9. Image Preparation and Analysis
2.10. Statistical Analysis
3. Results
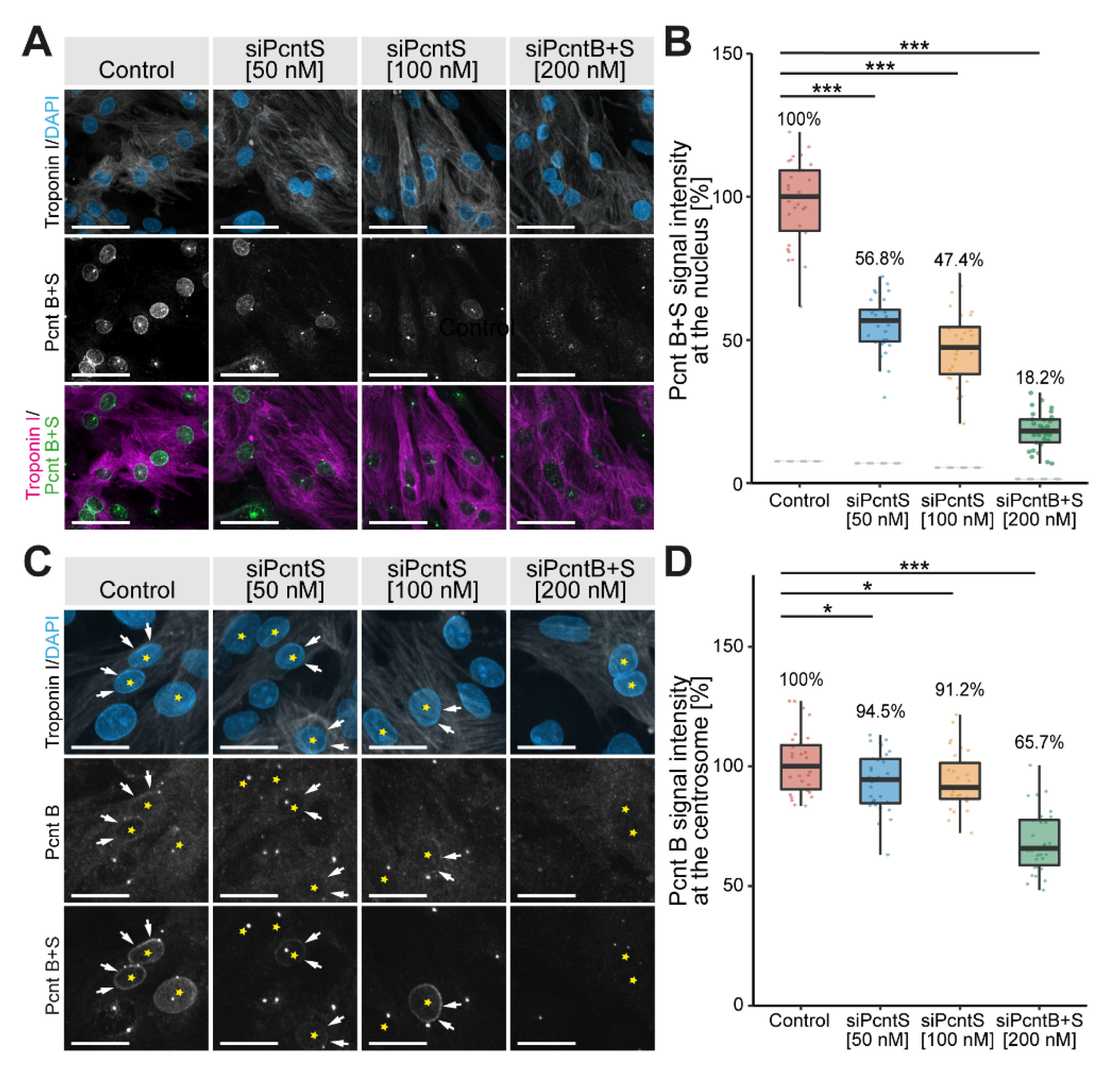
3.1. Pcnt B Preferentially Localizes at the Centrosome and Pcnt S at the Nuclear Envelope
3.2. Pcnt S Is Not Required for Centriole Cohesion
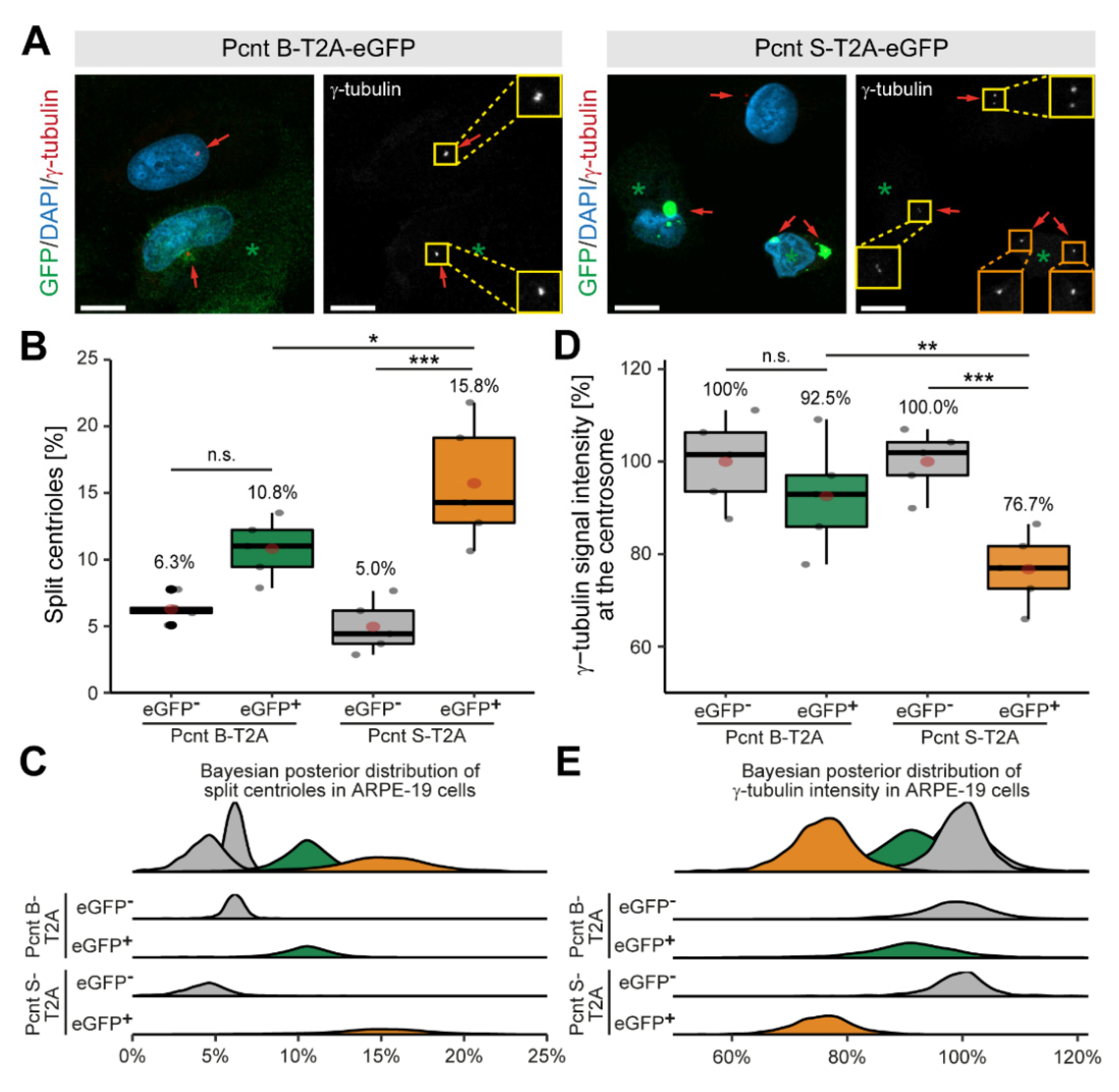
3.3. Ectopic Expression of Pcnt S Induces Centriole Splitting and Centriolar γ-Tubulin Reduction
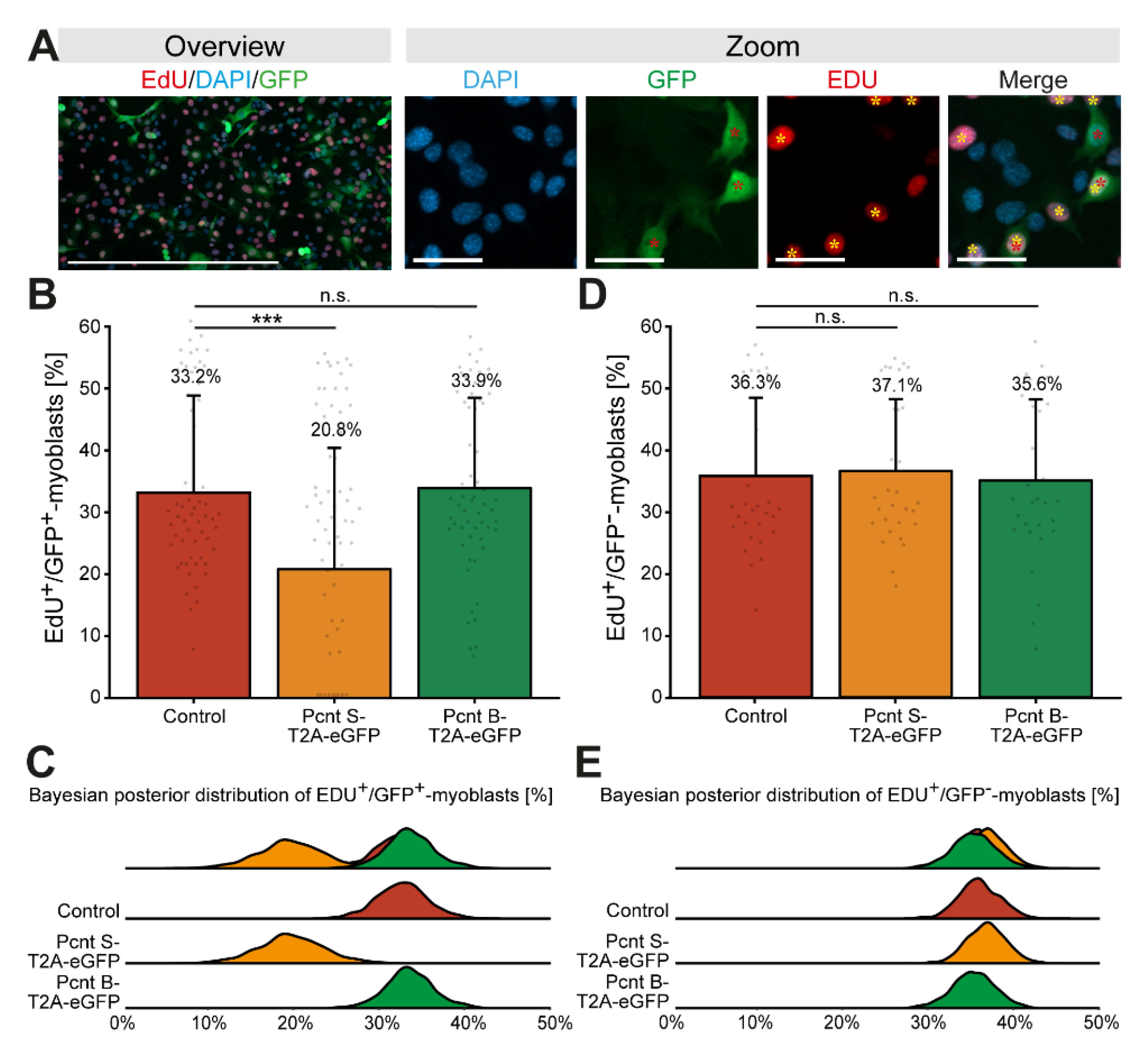
3.4. Ectopic Pcnt S Expression Impairs DNA Synthesis
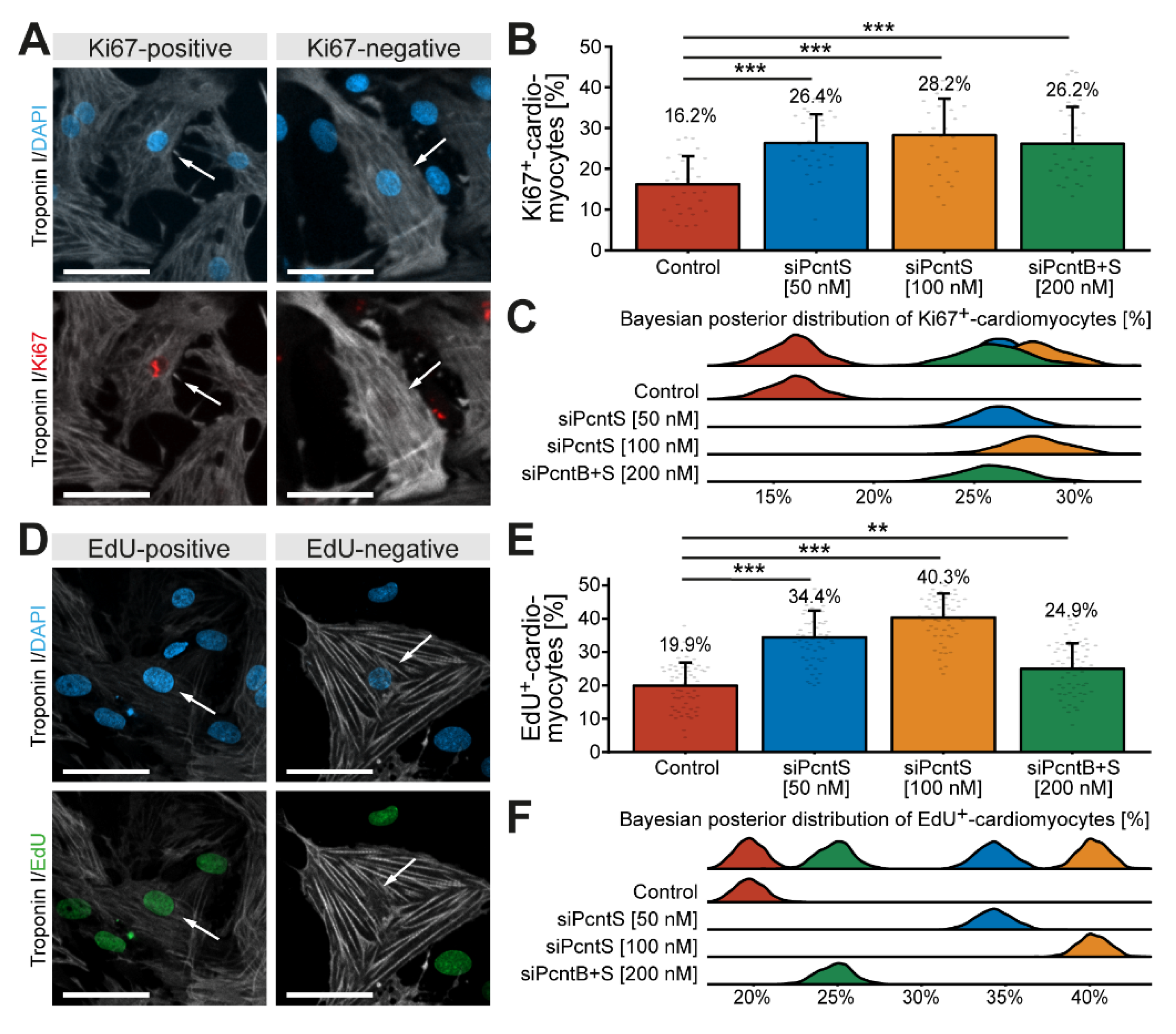
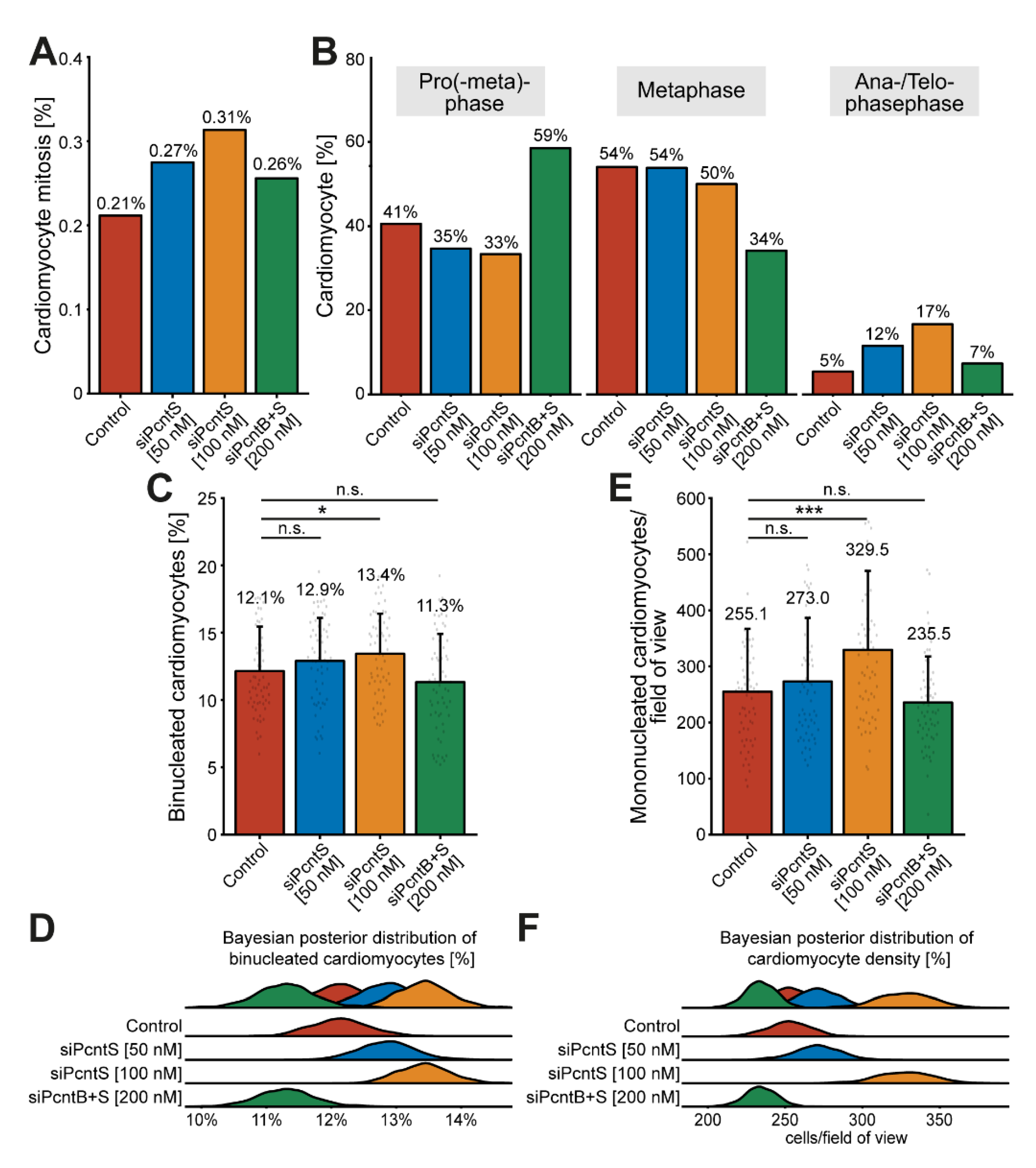
3.5. Pcnt S Depletion Enhances Serum-Induced Cell Cycle Re-Entry in Cardiomyocytes
3.6. Pcnt S Depletion Enhances Serum-Induced Cell Division in Cardiomyocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Bertero, A.; Murry, C.E. Hallmarks of cardiac regeneration. Nat. Rev. Cardiol. 2018, 15, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Rapid Transition of Cardiac Myocytes from Hyperplasia to Hypertrophy During Postnatal Development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. Heart Circ. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Engel, F.B. The Cardiomyocyte Cell Cycle in Hypertrophy, Tissue Homeostasis, and Regeneration. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 67–96. [Google Scholar] [CrossRef] [PubMed]
- Engel, F.B.; Schebesta, M.; Duong, M.T.; Lu, G.; Ren, S.; Madwed, J.B.; Jiang, H.; Wang, Y.; Keating, M.T. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005, 19, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, M.; Engel, F.B. Advances in heart regeneration based on cardiomyocyte proliferation and regenerative potential of binucleated cardiomyocytes and polyploidization. Clin. Sci. 2019, 133, 1229–1253. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Becker, R.; Engel, F.B. Towards regenerating the mammalian heart: Challenges in evaluating experimentally induced adult mammalian cardiomyocyte proliferation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1045–H1054. [Google Scholar] [CrossRef]
- Leone, M.; Engel, F.B. Pseudo-bipolar spindle formation and cell division in postnatal binucleated cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 134, 69–73. [Google Scholar] [CrossRef]
- Leone, M.; Musa, G.; Engel, F.B. Cardiomyocyte binucleation is associated with aberrant mitotic microtubule distribution, mislocalization of RhoA and IQGAP3, as well as defective actomyosin ring anchorage and cleavage furrow ingression. Cardiovasc. Res. 2018, 114, 1115–1131. [Google Scholar] [CrossRef]
- Leone, M.; Magadum, A.; Engel, F.B. Cardiomyocyte proliferation in cardiac development and regeneration: A guide to methodologies and interpretations. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1237–H1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Delaval, B.; Doxsey, S.J. Pericentrin in cellular function and disease. J. Cell Biol. 2010, 188, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Takao, D.; Ito, K.K.; Takahashi, M.; Kitagawa, D. The Cep57-pericentrin module organizes PCM expansion and centriole engagement. Nat. Commun. 2019, 10, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, K.; Asanuma, M.; Miyazaki, I.; Matsuzaki, S.; Tohyama, M.; Ogawa, N. Characterization of pericentrin isoforms in vivo. Biochem. Biophys. Res. Commun. 2006, 351, 745–749. [Google Scholar] [CrossRef]
- Flory, M.R.; Davis, T.N. The centrosomal proteins pericentrin and kendrin are encoded by alternatively spliced products of one gene. Genomics 2003, 82, 401–405. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Vergarajauregui, S.; Wu, C.-C.; Piatkowski, T.; Becker, R.; Leone, M.; Hirth, S.; Ricciardi, F.; Falk, N.; Giessl, A.; et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. eLife 2015, 4, e05563. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.; Leone, M.; Engel, F.B. Microtubule Organization in Striated Muscle Cells. Cells 2020, 9, 1395. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Becker, R.; Steffen, U.; Sharkova, M.; Esser, T.; Petzold, J.; Billing, F.; Kapiloff, M.S.; Schett, G.; Thievessen, I.; et al. AKAP6 orchestrates the nuclear envelope microtubule-organizing center by linking golgi and nucleus via AKAP9. eLife 2020, 9, e61669. [Google Scholar] [CrossRef] [PubMed]
- Galati, D.F.; Sullivan, K.D.; Pham, A.T.; Espinosa, J.M.; Pearson, C.G. Trisomy 21 Represses Cilia Formation and Function. Dev. Cell 2018, 46, 641–650.e6. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Rhee, K. Separase-dependent cleavage of pericentrin B is necessary and sufficient for centriole disengagement during mitosis. Cell Cycle 2012, 11, 2476–2485. [Google Scholar] [CrossRef] [Green Version]
- Mühlhans, J.; Brandstätter, J.H.; Giessl, A. The centrosomal protein pericentrin identified at the basal body complex of the connecting cilium in mouse photoreceptors. PLoS ONE 2011, 6, e26496. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; Lindquist, R.A.; Papallo, A.; Sabatini, D.M.; Golland, P.; Carpenter, A.E. CellProfiler Analyst: Data exploration and analysis software for complex image-based screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 27 April 2020).
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. R Package Version 3.1-152. 2021. Available online: https://CRAN.R-project.org/package=nlme (accessed on 27 April 2020).
- Morey, R.D.; Rouder, J.N.; Jamil, T. BayesFactor: Computation of Bayes Factors for Common Designs (Version 0.9.12-2). 2015. Available online: https://cran.r-project.org/web/packages/BayesFactor/BayesFactor.pdf (accessed on 27 April 2020).
- Doxsey, S.J.; Stein, P.; Evans, L.; Calarco, P.D.; Kirschner, M. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell 1994, 76, 639–650. [Google Scholar] [CrossRef]
- Kim, S.; Rhee, K. Importance of the CEP215-pericentrin interaction for centrosome maturation during mitosis. PLoS ONE 2014, 9, e87016. [Google Scholar] [CrossRef] [Green Version]
- van Amerongen, M.J.; Engel, F.B. Features of cardiomyocyte proliferation and its potential for cardiac regeneration. J. Cell. Mol. Med. 2008, 12, 2233–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimpel, P.; Lee, Y.L.; Sobota, R.M.; Calvi, A.; Koullourou, V.; Patel, R.; Mamchaoui, K.; Nedelec, F.; Shackleton, S.; Schmoranzer, J.; et al. Nesprin-1alpha-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Curr. Biol. 2017, 27, 2999–3009.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.C.; Gatter, K.C. Ki67 protein: The immaculate deception? Histopathology 2002, 40, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Meckert, P.; Rivello, H.; Vigliano, C.; Gonzalez, P.; Favaloro, R.; Laguens, R. Endomitosis and polyploidization of myocardial cells in the periphery of human acute myocardial infarction. Cardiovasc. Res. 2005, 67, 116–123. [Google Scholar] [CrossRef]
- Muhlhans, J.; Giessl, A. Pericentrin in health and disease: Exploring the patchwork of Pericentrin splice variants. Commun. Integr. Biol. 2012, 5, 304–307. [Google Scholar] [CrossRef]
- Sepulveda, G.; Antkowiak, M.; Brust-Mascher, I.; Mahe, K.; Ou, T.; Castro, N.M.; Christensen, L.N.; Cheung, L.; Jiang, X.; Yoon, D.; et al. Co-translational protein targeting facilitates centrosomal recruitment of PCNT during centrosome maturation in vertebrates. eLife 2018, 7, e34959. [Google Scholar] [CrossRef] [PubMed]
- Mikule, K.; Delaval, B.; Kaldis, P.; Jurcyzk, A.; Hergert, P.; Doxsey, S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 2007, 9, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinfeldt, J.; Becker, R.; Vergarajauregui, S.; Engel, F.B. Alternative Splicing of Pericentrin Contributes to Cell Cycle Control in Cardiomyocytes. J. Cardiovasc. Dev. Dis. 2021, 8, 87. https://doi.org/10.3390/jcdd8080087
Steinfeldt J, Becker R, Vergarajauregui S, Engel FB. Alternative Splicing of Pericentrin Contributes to Cell Cycle Control in Cardiomyocytes. Journal of Cardiovascular Development and Disease. 2021; 8(8):87. https://doi.org/10.3390/jcdd8080087
Chicago/Turabian StyleSteinfeldt, Jakob, Robert Becker, Silvia Vergarajauregui, and Felix B. Engel. 2021. "Alternative Splicing of Pericentrin Contributes to Cell Cycle Control in Cardiomyocytes" Journal of Cardiovascular Development and Disease 8, no. 8: 87. https://doi.org/10.3390/jcdd8080087
APA StyleSteinfeldt, J., Becker, R., Vergarajauregui, S., & Engel, F. B. (2021). Alternative Splicing of Pericentrin Contributes to Cell Cycle Control in Cardiomyocytes. Journal of Cardiovascular Development and Disease, 8(8), 87. https://doi.org/10.3390/jcdd8080087