
Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine

 and
and 
Abstract
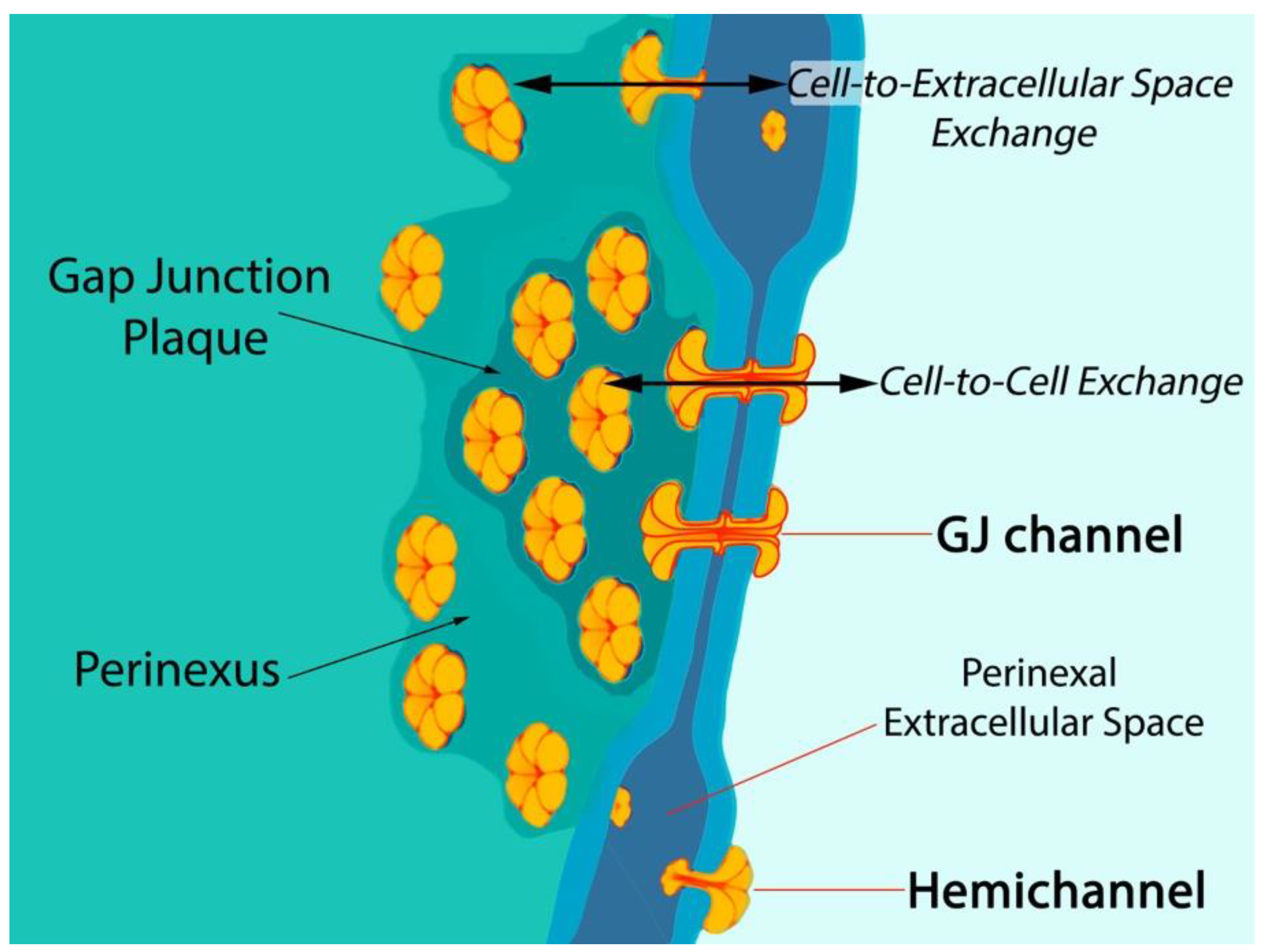
1. Connexin43-Formed Channels
2. Connexin43 and Myocardial Pathology
3. Cx43-Targeting Therapeutic Peptides
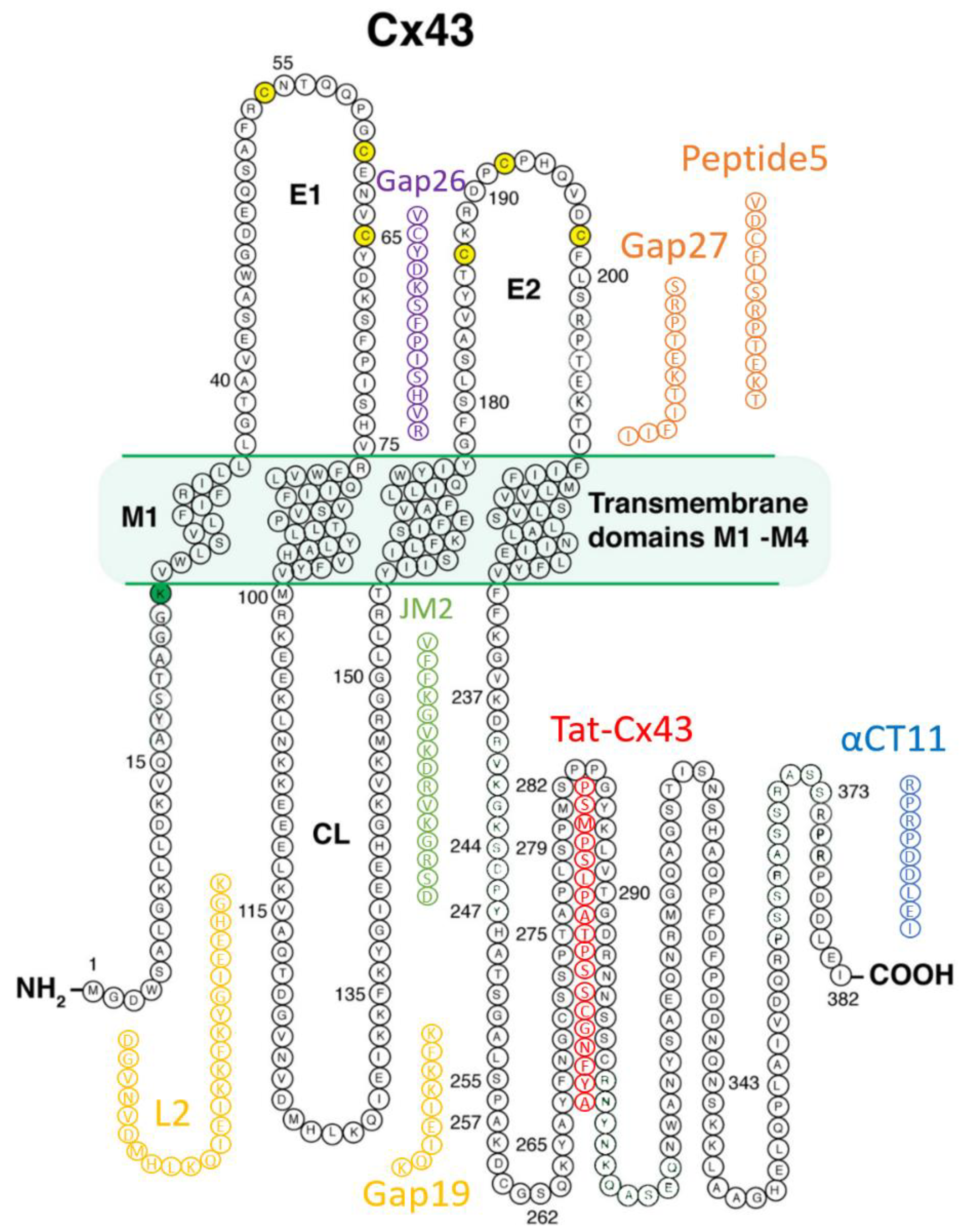
3.1. Mimetic Peptides Based on Cx43 Extracellular Domains
3.1.1. Gap26 and Gap27
3.1.2. Peptide5
3.2. Mimetic Peptides Based on Cx43 Cytoplasmic Loop Domains
Gap19 and L2
3.3. Mimetic Peptides Based on Cx43 Cytoplasmic Terminal Domain
3.3.1. alphaCT/αCT Peptides
3.3.2. CT9
3.3.3. Other Cx43 CT Mimetic or Targeting Peptides
4. Barriers to Clinical Translation of Cx43-Targeting Peptidic Therapeutics
4.1. Stability of Peptidic Drugs In Vivo
4.2. Cell and Tissue Penetration of Peptidic Drugs
4.3. The Immune System and Peptidic Drugs
4.4. The Potential of Extracellular Vesicles as Vehicles for Mimetic Peptides
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2017, 10, a029348. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef]
- Harris, A.L. Electrical coupling and its channels. J. Gen. Physiol. 2018, 150, 1606–1639. [Google Scholar] [CrossRef]
- Gourdie, R.; Smyth, J.; Poelzing, S. Gap Junctional Connexin43: Novel Insights from the New Millennium and Their Clinical Implications. In Cardiac Electrophysiology: From Cell to Bedside, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein(-)Protein Interactions with Connexin43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.T.; Santos, W.; Poelzing, S.; Gourdie, R.G. The role of the gap junction perinexus in cardiac conduction: Potential as a novel anti-arrhythmic drug target. Prog. Biophys. Mol. Biol. 2019, 144, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225. [Google Scholar] [CrossRef]
- Laird, D.W.; Puranam, K.L.; Revel, J.P. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991, 273 Pt 1, 67–72. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef]
- John, S.A.; Kondo, R.; Wang, S.-Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kam, Y.; Koo, S.K.; Joe, C.O. Gating Connexin43 channels reconstituted in lipid vesicles by mitogen-activated protein kinase phosphorylation. J. Biol. Chem. 1999, 274, 5581–5587. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Orellana, J.A.; Bennett, M.V.L.; Sáez, J.C. Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia-reperfusion. J. Membr. Biol. 2007, 218, 49–63. [Google Scholar] [CrossRef]
- De Vuyst, E.; Wang, N.; Decrock, E.; De Bock, M.; Vinken, M.; Van Moorhem, M.; Lai, C.; Culot, M.; Rogiers, V.; Cecchelli, R.; et al. Ca (2+ ) regulation of Connexin43 hemichannels in C6 glioma and glial cells. Cell Calcium 2009, 46, 176–187. [Google Scholar] [CrossRef]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. 1), 11. [Google Scholar] [CrossRef]
- Ek-Vitorín, J.F.; Pontifex, T.K.; Burt, J.M. Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. Int. J. Mol. Sci. 2018, 19, 1659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bai, X.; Liu, Y.; Wang, K.; Shen, B.; Sun, X. Current Concepts and Perspectives on Connexin43: A Mini Review. Curr. Protein Pept. Sci. 2018, 19, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.E.; Gourdie, R.G. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Rhett, J.M.; Ghatnekar, G.S. Cardiac to cancer: Connecting connexins to clinical opportunity. FEBS Lett. 2014, 588, 1349–1364. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Green, C.R.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannels: A potential drug target for the treatment of diabetic retinopathy. Drug Discov. Today 2019, 24, 1627–1636. [Google Scholar] [CrossRef]
- Mugisho, O.O.; Rupenthal, I.D.; Paquet-Durand, F.; Acosta, M.L.; Green, C.R. Targeting connexin hemichannels to control the inflammasome: The correlation between connexin43 and NLRP3 expression in chronic eye disease. Expert Opin. Ther. Targets 2019, 23, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, T.; Vandenabeele, P.; Bultynck, G.; Leybaert, L.; Krysko, D.V. Therapeutic Targeting of Connexin Channels: New Views and Challenges. Trends Mol. Med. 2018, 24, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Cocozzelli, A.G.; White, T.W. Connexin43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. Int. J. Mol. Sci. 2019, 20, 6186. [Google Scholar] [CrossRef]
- Varela-Vázquez, A.; Guitián-Caamaño, A.; Carpintero-Fernandez, P.; Fonseca, E.; Sayedyahossein, S.; Aasen, T.; Penuela, S.; Mayán, M.D. Emerging functions and clinical prospects of connexins and pannexins in melanoma. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188380. [Google Scholar] [CrossRef]
- Kieken, F.; Mutsaers, N.; Dolmatova, E.; Virgil, K.; Wit, A.L.; Kellezi, A.; Hirst-Jensen, B.J.; Duffy, H.S.; Sorgen, P.L. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 2009, 104, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.H.; Green, C.R.; Peters, N.S.; Rothery, S.; Severs, N.J. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 1991, 139, 801–821. [Google Scholar] [PubMed]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; King, T.J.; Heyman, N.S.; Lampe, P.D.; Burt, J.M. Selectivity of Connexin43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 2006, 98, 1498–1505. [Google Scholar] [CrossRef]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Bao, X.; Altenberg, G.A.; Reuss, L. Mechanism of regulation of the gap junction protein Connexin43 by protein kinase C-mediated phosphorylation. Am. J. Physiol. Cell Physiol. 2004, 286, C647–C654. [Google Scholar] [CrossRef]
- Boengler, K.; Stahlhofen, S.; Van De Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of Connexin43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef]
- Totland, M.Z.; Rasmussen, N.L.; Knudsen, L.M.; Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: Physiological and pathophysiological implications. Cell Mol Life Sci. 2020, 77, 573–591. [Google Scholar] [CrossRef]
- Laing, J.G.; Beyer, E.C. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 1995, 270, 26399–26403. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Girao, H.; Yuste, A.; Patel, B.; Marques, C.; Spray, D.C.; Pereira, P.; Cuervo, A.M. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012, 23, 2156–2169. [Google Scholar] [CrossRef]
- Leithe, E.; Kjenseth, A.; Sirnes, S.; Stenmark, H.; Brech, A.; Rivedal, E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci. 2009, 122, 3883–3893. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef] [PubMed]
- Zu, L.; Wen, N.; Liu, C.; Zhao, M.; Zheng, L. Connexin43 and Myocardial Ischemia-Reperfusion Injury. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 14–16. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef]
- Ramadan, R.; Baatout, S.; Aerts, A.; Leybaert, L. The role of connexin proteins and their channels in radiation-induced atherosclerosis. Cell Mol Life Sci. 2021, 78, 3087–3103. [Google Scholar] [CrossRef]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front Oncol. 2018, 8, 646. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 12, 775–788. [Google Scholar] [CrossRef]
- Farah, A.; Barbagelata, A. Unmet goals in the treatment of Acute Myocardial Infarction: Review. F1000Res 2017, 6, F1000 Faculty Rev-1243. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.C.; Williams, O.J.; Martin, P.E.; Evans, W.H. ATP release by cardiac myocytes in a simulated ischaemia model: Inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 2009, 605, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Soleymani, S.; Madakshire, R.; Insel, P.A. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 2012, 26, 2580–2591. [Google Scholar] [CrossRef]
- Dosch, M.; Zindel, J.; Jebbawi, F.; Melin, N.; Sanchez-Taltavull, D.; Stroka, D.; Candinas, D.; Beldi, G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. Elife 2019, 8, 2670. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.M.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Gehmlich, K.; Syrris, P.; Reimann, M.; Asimaki, A.; Ehler, E.; Evans, A.; Quarta, G.; Pantazis, A.; Saffitz, J.E.; McKenna, W.J. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc. Pathol. 2012, 21, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.; Asimaki, A.; van Rijen, H.V.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.; et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013, 10, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G. Towbin JA and Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. Dystrophin and the membrane skeleton. Curr. Opin. Cell Biol. 1993, 5, 82–87. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Connor, E. Clinical outcome measures for trials in Duchenne muscular dystrophy: Report from International Working Group meetings. Clin. Investig. 2011, 1, 1217–1235. [Google Scholar] [CrossRef]
- Shaw, R.M.; Saffitz, J.E. A role for connexin-43 in Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1608–1610. [Google Scholar] [CrossRef]
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.-H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, e130091. [Google Scholar] [CrossRef]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Gonzalez, J.P.; Zhao, Q.; Xie, L.-H.; Li, H.; Liu, T.; Wehrens, X.H.; Lampe, P.D.; et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1727. [Google Scholar] [CrossRef]
- Vielma, A.Z.; Boric, M.P.; Gonzalez, D.R. Apocynin Treatment Prevents Cardiac Connexin43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. Int. J. Mol. Sci. 2020, 21, 5415. [Google Scholar] [CrossRef]
- Nouet, J.; Himelman, E.; Lahey, K.C.; Zhao, Q.; Fraidenraich, D. Connexin-43 reduction prevents muscle defects in a mouse model of manifesting Duchenne muscular dystrophy female carriers. Sci. Rep. 2020, 10, 5683. [Google Scholar] [CrossRef]
- Borin, D.; Peña, B.; Chen, S.N.; Long, C.S.; Taylor, M.R.; Mestroni, L.; Sbaizero, O. Altered microtubule structure, hemichannel localization and beating activity in cardiomyocytes expressing pathologic nuclear lamin A/C. Heliyon 2020, 6, e03175. [Google Scholar] [CrossRef]
- Iyyathurai, J.; D’Hondt, C.; Wang, N.; De Bock, M.; Himpens, B.; Retamal, M.A.; Stehberg, J.; Leybaert, L.; Bultynck, G. Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: Gap junctions versus hemichannels. Neuropharmacology 2013, 75, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Faniku, C.; O’Shaughnessy, E.; Lorraine, C.; Johnstone, S.R.; Graham, A.; Greenhough, S.; Martin, P.E.M. The Connexin Mimetic Peptide Gap27 and Cx43-Knockdown Reveal Differential Roles for Connexin43 in Wound Closure Events in Skin Model Systems. Int. J. Mol. Sci. 2018, 19, 604. [Google Scholar] [CrossRef] [PubMed]
- Cooreman, A.; Van Campenhout, R.; Ballet, S.; Annaert, P.; Bossche, B.V.D.; Colle, I.; Cogliati, B.; Vinken, M. Connexin and Pannexin (Hemi)Channels: Emerging Targets in the Treatment of Liver Disease. Hepatology 2019, 69, 1317–1323. [Google Scholar] [CrossRef]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflugers Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.-O. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Hawat, G.; Hélie, P.; Baroudi, G. Single intravenous low-dose injections of Connexin43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J. Mol. Cell Cardiol. 2012, 53, 559–566. [Google Scholar] [CrossRef]
- Behmenburg, F.; Pickert, E.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Berger, M.M. The Cardioprotective Effect of Dexmedetomidine in Rats Is Dose-Dependent and Mediated by BKCa Channels. J. Cardiovasc. Pharmacol. 2017, 69, 228–235. [Google Scholar] [CrossRef]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 6878. [Google Scholar] [CrossRef]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef]
- Boengler, K.; Ungefug, E.; Heusch, G.; Leybaert, L.; Schulz, R. Connexin43 impacts on mitochondrial potassium uptake. Front. Pharmacol. 2013, 4, 73. [Google Scholar] [CrossRef]
- Gonzalez, J.P.; Ramachandran, J.; Xie, L.H.; Contreras, J.E.; Fraidenraich, D. Selective Connexin43 Inhibition Prevents Isoproterenol-Induced Arrhythmias and Lethality in Muscular Dystrophy Mice. Sci. Rep. 2015, 5, 13490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of alpha Carboxyl Terminus 1 Peptide with the Connexin43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Jiang, J.; Abbate, A.; Jourdan, J.L.; Gourdie, R.G. Abstract 13803: A Short Connexin43 Carboxyl Terminal-Based Peptide Permeates Hemichannels and Provides Post-Infarction Cardioprotection in vivo. Circ. Res. 2019, 140, A13803. [Google Scholar]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell. 2005, 16, 5686–5698. [Google Scholar] [CrossRef]
- Becker, D.L.; Evans, W.H.; Green, C.R.; Warner, A. Functional analysis of amino acid sequences in connexin43 involved in intercellular communication through gap junctions. J. Cell Sci. 1995, 108 Pt 4, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Chaytor, A.T.; Evans, W.H.; Griffith, T.M. Peptides homologous to extracellular loop motifs of Connexin43 reversibly abolish rhythmic contractile activity in rabbit arteries. J. Physiol. 1997, 503 Pt 1, 99–110. [Google Scholar] [CrossRef]
- Desplantez, T.; Verma, V.; Leybaert, L.; Evans, W.H.; Weingart, R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol. Res. 2012, 65, 546–552. [Google Scholar] [CrossRef]
- Warner, A.; Clements, D.K.; Parikh, S.; Evans, W.H.; DeHaan, R.L. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 1995, 488 Pt 3, 721–728. [Google Scholar] [CrossRef]
- Berthoud, V.M.; Beyer, E.C.; Seul, K.H. Peptide inhibitors of intercellular communication. Am J Physiol Lung Cell Mol Physiol. 2000, 279, 619–622. [Google Scholar] [CrossRef]
- Boitano, S.; Evans, W.H. Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L623–L630. [Google Scholar] [CrossRef]
- Liu, F.; Arce, F.T.; Ramachandran, S.; Lal, R. Nanomechanics of hemichannel conformations: Connexin flexibility underlying channel opening and closing. J. Biol. Chem. 2006, 281, 23207–23217. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Griffin, J.M.; Harris, P.W.; Chan, S.H.C.; Nicholson, L.F.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular Connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Ujiie, H.; Chaytor, A.T.; Bakker, L.M.; Griffith, T.M. Essential role of Gap junctions in NO- and prostanoid-independent relaxations evoked by acetylcholine in rabbit intracerebral arteries. Stroke 2003, 34, 544–550. [Google Scholar] [CrossRef][Green Version]
- Sandow, S.L.; Goto, K.; Rummery, N.M.; Hill, C.E. Developmental changes in myoendothelial gap junction mediated vasodilator activity in the rat saphenous artery. J. Physiol. 2004, 556, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Haddock, R.E.; Grayson, T.H.; Brackenbury, T.D.; Meaney, K.R.; Neylon, C.B.; Sandow, S.L.; Hill, C.E. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2047–H2056. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Inoue, T.; Kanno, Y.; Okada, H.; Hill, C.E.; Suzuki, H. Connexins 37 and 40 transduce purinergic signals mediating renal autoregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1–R11. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Platoshyn, O.; Suárez, J.; Yuan, J.X.-J.; Dillmann, W.H. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am. J. Physiol. Cell Physiol. 2008, 295, C221–C230. [Google Scholar] [CrossRef]
- Cotter, M.L.; Boitano, S.; Lampe, P.D.; Solan, J.L.; Vagner, J.; Ek-Vitorin, J.F.; Burt, J.M. The lipidated connexin mimetic peptide SRPTEKT-Hdc is a potent inhibitor of Cx43 channels with specificity for the pS368 phospho-isoform. Am. J. Physiol. Cell Physiol. 2019, 317, C825–C842. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.B.; Green, C.R. Connexin43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef]
- Acosta, M.L.; Nor, M.N.M.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin therapeutics: Blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef]
- Mugisho, O.O.; Green, C.R.; Squirrell, D.M.; Bould, S.; Danesh-Meyer, H.V.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. J. Mol. Med. 2019, 97, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.; Green, C.R.; Rupenthal, I.D.; Mugisho, O.O. Connexin43 hemichannel block protects against retinal pigment epithelial cell barrier breakdown. Acta Diabetol. 2020, 57, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Price, G.W.; Chadjichristos, C.E.; Kavvadas, P.; Tang, S.C.W.; Yiu, W.H.; Green, C.R.; Potter, J.A.; Siamantouras, E.; Squires, P.E.; Hills, C.E. Blocking Connexin-43 mediated hemichannel activity protects against early tubular injury in experimental chronic kidney disease. Cell Commun. Signal. 2020, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Cassara, C.; Lin, X.; Veenstra, R.D. Calcium-calmodulin gating of a pH-insensitive isoform of connexin43 gap junctions. Biochem. J. 2019, 476, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef]
- Lissoni, A.; Wang, N.; Nezlobinskii, T.; De Smet, M.; Panfilov, A.V.; Vandersickel, N.; Leybaert, L.; Witschas, K. Gap19, a Cx43 Hemichannel Inhibitor, Acts as a Gating Modifier That Decreases Main State Opening While Increasing Substate Gating. Int. J. Mol. Sci. 2020, 21, 7340. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Wang, N.; Bechberger, J.F.; De Bock, M.; Lampe, P.D.; Leybaert, L.; Naus, C.C. Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 2019, 216, 916–935. [Google Scholar] [CrossRef]
- Coutinho, F.P.; Green, C.R.; Acosta, M.L.; Rupenthal, I.D. Xentry-Gap19 inhibits Connexin43 hemichannel opening especially during hypoxic injury. Drug Deliv. Transl. Res. 2020, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, R.; Vromans, E.; Anang, D.C.; Goetschalckx, I.; Hoorelbeke, D.; Decrock, E.; Baatout, S.; Leybaert, L.; Aerts, A. Connexin43 Hemichannel Targeting With TAT-Gap19 Alleviates Radiation-Induced Endothelial Cell Damage. Front. Pharmacol. 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- D’Hondt, C.; Iyyathurai, J.; Vinken, M.; Rogiers, V.; Leybaert, L.; Himpens, B.; Bultynck, G. Regulation of connexin- and pannexin-based channels by post-translational modifications. Biol. Cell 2013, 105, 373–398. [Google Scholar] [CrossRef]
- Spagnol, G.; Al-Mugotir, M.; Kopanic, J.L.; Zach, S.; Li, H.; Trease, A.J.; Stauch, K.L.; Grosely, R.; Cervantes, M.; Sorgen, P.L.; et al. Secondary structural analysis of the carboxyl-terminal domain from different connexin isoforms. Biopolymers 2016, 105, 143–162. [Google Scholar] [CrossRef] [PubMed]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef]
- Moore, K.; Bryant, Z.J.; Ghatnekar, G.; Singh, U.P.; Gourdie, R.G.; Potts, J.D. A synthetic Connexin43 mimetic peptide augments corneal wound healing. Exp. Eye Res. 2013, 115, 178–188. [Google Scholar] [CrossRef]
- Grek, C.L.; Prasad, G.M.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Ghatnekar, G.S. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015, 23, 203–212. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A Multicenter Randomized Controlled Trial Evaluating a Cx43-Mimetic Peptide in Cutaneous Scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018, 20, e121900. [Google Scholar] [CrossRef] [PubMed]
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for Connexin43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, T.; De Smet, M.A.J.; Verwaerde, S.; Decrock, E.; Czekaj, A.; Vandenbroucke, R.E.; Lemeire, K.; Goncalves, A.; Declercq, W.; Vandenabeele, P.; et al. Blocking connexin43 hemichannels protects mice against tumour necrosis factor-induced inflammatory shock. Sci. Rep. 2019, 9, 16623. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. Enhanced PKCepsilon mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels 2011, 5, 236–240. [Google Scholar] [CrossRef]
- Calder, B.W.; Matthew Rhett, J.; Bainbridge, H.; Fann, S.A.; Gourdie, R.G.; Yost, M.J. Inhibition of Connexin43 hemichannel-mediated ATP release attenuates early inflammation during the foreign body response. Tissue Eng. Part A 2015, 21, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef]
- Lamouille, S.; Smyth, J.W.; O’Rourke, L.; Kanabur, P.; Guo, S.; Jourdan, L.J.; Sheng, Z.; Gourdie, R.G. Targeting glioblastoma cancer stem cells with a novel Connexin43 mimetic peptide. Cancer Res. 2020, 77, 4765. [Google Scholar]
- Pelaz, S.G.; Jaraíz-Rodríguez, M.; Álvarez-Vázquez, A.; Talaverón, R.; García-Vicente, L.; Flores-Hernández, R.; De Cedrón, M.G.; Tabernero, M.; De Molina, A.R.; Lillo, C.; et al. Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266-283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef]
- Shibayama, J.; Lewandowski, R.; Kieken, F.; Coombs, W.; Shah, S.; Sorgen, P.L.; Taffet, S.M.; Delmar, M. Identification of a novel peptide that interferes with the chemical regulation of connexin43. Circ. Res. 2006, 98, 1365–1372. [Google Scholar] [CrossRef]
- Verma, V.; Larsen, B.D.; Coombs, W.; Lin, X.; Spagnol, G.; Sorgen, P.L.; Taffet, S.M.; Delmar, M. Novel Pharmacophores of Connexin43 Based on the “RXP” Series of Cx43-Binding Peptides. Circ. Res. 2009, 105, 176–184. [Google Scholar] [CrossRef]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.-W. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: The action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 381, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Gottwald, M.; Tudyka, T.; Linke, W.; Klaus, W.; Dhein, S. Increase in gap junction conductance by an antiarrhythmic peptide. Eur. J. Pharmacol. 1997, 327, 65–72. [Google Scholar] [CrossRef]
- Xing, D.; Kjolbye, A.L.; Nielsen, M.S.; Petersen, J.S.; Harlow, K.W.; Holstein-Rathlou, N.H.; Martins, J.B. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J. Cardiovasc. Electrophysiol. 2003, 14, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Keith, J.C.; Schaub, R.G.; Smith, R.P.; Feldman, H.S.; Haugan, K.; Kantrowitz, J.; Wang, P.J.; et al. Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J. Pharmacol. Exp. Ther. 2006, 317, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-S.; Zhong, J.-Q.; Zeng, Q.-X.; Liu, H.-Z.; Su, G.-Y.; Zhang, Y. Effect of ZP123, a gap junction modifier, on prolonged ventricular fibrillation in swine. Cardiology 2011, 118, 147–152. [Google Scholar] [CrossRef]
- Skyschally, A.; Walter, B.; Hansen, R.S.; Heusch, G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 383–391. [Google Scholar] [CrossRef]
- Reynolds, J. Zealand announces results of a Phase II Proof-of-Concept trial with danegaptide for cardiac reperfusion injuries. Fierce Biotech 2016. Available online: https://www.fiercebiotech.com/biotech/zealand-announces-results-of-a-phase-ii-proof-of-concept-trial-danegaptide-for-cardiac (accessed on 1 March 2021).
- ClinicalTrials.gov. Gap Junction Potentiation of Endothelial Function with Rotigaptide in the Human Forearm Arterial Circulation—Effects of Ischaemia Induced Endothelial Dysfunction. 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT00901563?term=rotigaptide&draw=2&rank=1 (accessed on 1 March 2021).
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Bliss, M. Banting’s, Best’s, and Collip’s accounts of the discovery of insulin. Bull. Hist. Med. 1982, 56, 554–568. [Google Scholar]
- Chang, S.-G.; Choi, K.-D.; Jang, S.-H.; Shin, H.-C. Role of disulfide bonds in the structure and activity of human insulin. Mol. Cells 2003, 16, 323–330. [Google Scholar] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.A.; Deber, C.M. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2017; 1859, 577–585. [Google Scholar]
- Moore, K.; Ghatnekar, G.; Gourdie, R.G.; Potts, J.D. Impact of the controlled release of a Connexin43 peptide on corneal wound closure in an STZ model of type I diabetes. PLoS ONE 2014, 9, e86570. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Amos, J.; Davis, J.; Gourdie, R.; Potts, J.D. Characterization of polymeric microcapsules containing a low molecular weight peptide for controlled release. Microsc. Microanal. 2013, 19, 213–226. [Google Scholar] [CrossRef]
- Roberts, R.; Smyth, J.W.; Will, J.; Roberts, P.; Grek, C.L.; Ghatnekar, G.S.; Sheng, Z.; Gourdie, R.G.; Lamouille, S.; Foster, E.J. Development of PLGA nanoparticles for sustained release of a connexin43 mimetic peptide to target glioblastoma cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 108, 110191. [Google Scholar] [CrossRef]
- Miner-Williams, W.M.; Stevens, B.R.; Moughan, P.J. Are intact peptides absorbed from the healthy gut in the adult human? Nutr. Res. Rev. 2014, 27, 308–329. [Google Scholar] [CrossRef]
- Fu, C.; Xiang, Y.; Li, X.; Fu, A. Targeted transport of nanocarriers into brain for theranosis with rabies virus glycoprotein-derived peptide. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 87, 155–166. [Google Scholar] [CrossRef]
- Neijssen, J.; Herberts, C.; Drijfhout, J.W.; Reits, E.A.J.; Janssen, L.; Neefjes, J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature 2005, 434, 83–88. [Google Scholar] [CrossRef]
- Yang, J.; Luo, Y.; Shibu, M.A.; Toth, I.; Skwarczynskia, M.; Skwarczynski, M.; Skwarczyski, M. Cell-penetrating Peptides: Efficient Vectors for Vaccine Delivery. Curr. Drug Deliv. 2019, 16, 430–443. [Google Scholar] [CrossRef]
- Pinheiro, A.; Silva, A.M.; Teixeira, J.H.; Goncalves, R.M.; Almeida, M.I.; Barbosa, M.A.; Santos, S.G. Extracellular vesicles: Intelligent delivery strategies for therapeutic applications. J. Control. Release 2018, 289, 56–69. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ma, J.; Sun, T.; Zhou, Q.; Wang, W.; Wang, G.; Wu, P.; Wang, H.; Jiang, L.; et al. Exosomal circRNAs: Biogenesis, effect and application in human diseases. Mol. Cancer 2019, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Antes, T.J.; Middleton, R.C.; Luther, K.M.; Ijichi, T.; Peck, K.A.; Liu, W.J.; Valle, J.; Echavez, A.K.; Marbán, E. Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. J. Nanobiotechnol. 2018, 16, 61. [Google Scholar] [CrossRef]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef]
- Kishore, R.; Khan, M. More Than Tiny Sacks: Stem Cell Exosomes as Cell-Free Modality for Cardiac Repair. Circ. Res. 2016, 118, 330–343. [Google Scholar] [CrossRef]
- Sanwlani, R.; Fonseka, P.; Chitti, S.V.; Mathivanan, S. Milk-Derived Extracellular Vesicles in Inter-Organism, Cross-Species Communication and Drug Delivery. Proteomes 2020, 8, 11. [Google Scholar] [CrossRef]
- Galley, J.D.; Besner, G.E. The Therapeutic Potential of Breast Milk-Derived Extracellular Vesicles. Nutrients 2020, 12, 745. [Google Scholar] [CrossRef]
- Zempleni, J.; Sukreet, S.; Zhou, F.; Wu, D.; Mutai, E. Milk-Derived Exosomes and Metabolic Regulation. Annu. Rev. Anim. Biosci. 2019, 7, 245–262. [Google Scholar] [CrossRef]
- Manca, S.; Upadhyaya, B.; Mutai, E.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R.; Zempleni, J. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 2018, 8, 11321. [Google Scholar] [CrossRef]
- Epifantseva, I.; Xiao, S.; Baum, R.E.; Kléber, A.G.; Hong, T.; Shaw, R.M. An Alternatively Translated Connexin43 Isoform, GJA1-11k, Localizes to the Nucleus and Can Inhibit Cell Cycle Progression. Biomolecules 2020, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Flores, J.A.; Haddad, B.G.; Dolan, K.A.; Myers, J.B.; Yoshioka, C.C.; Copperman, J.; Zuckerman, D.M.; Reichow, S.L. Connexin-46/50 in a dynamic lipid environment resolved by CryoEM at 1.9 Å. Nat. Commun. 2020, 11, 4331. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Peptide Name | Type | Sequence | Effects in Disease Models Pertinent to Heart |
|---|---|---|---|
| Gap26 | Extracellular Loop | VCYDKSFPISJVR | Improves myocyte viability postischemia in vitro [46,75,76,77] |
| Gap27 | Extracellular Loop | SRPTEKTIFII | Reduces cardiac I/R injury severity ex vivo and in vivo [77,78,79] |
| L2 | Cytoplasmic Loop | DGVNVDMHLKQIEIKKFKYGIEEHGK | Protects myocytes against I/R injury [80] |
| Gap19 | Cytoplasmic Loop | KQIEIKKFK | Reduces cardiac I/R injury severity in vivo [80,81,82] |
| αCT1 | Cytoplasmic Terminus | Ant-RPRPDDLEI | Preischemic treatment decreases cardiac I/R injury severity ex vivo. In clinical testing in humans [26,83] |
| αCT11 | Cytoplasmic Terminus | RPRPDDLEI | Reduces cardiac I/R injury ex vivo and in vivo [83,84,85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsh, S.R.; Williams, Z.J.; Pridham, K.J.; Gourdie, R.G. Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine. J. Cardiovasc. Dev. Dis. 2021, 8, 52. https://doi.org/10.3390/jcdd8050052
Marsh SR, Williams ZJ, Pridham KJ, Gourdie RG. Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine. Journal of Cardiovascular Development and Disease. 2021; 8(5):52. https://doi.org/10.3390/jcdd8050052
Chicago/Turabian StyleMarsh, Spencer R., Zachary J. Williams, Kevin J. Pridham, and Robert G. Gourdie. 2021. "Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine" Journal of Cardiovascular Development and Disease 8, no. 5: 52. https://doi.org/10.3390/jcdd8050052
APA StyleMarsh, S. R., Williams, Z. J., Pridham, K. J., & Gourdie, R. G. (2021). Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine. Journal of Cardiovascular Development and Disease, 8(5), 52. https://doi.org/10.3390/jcdd8050052