
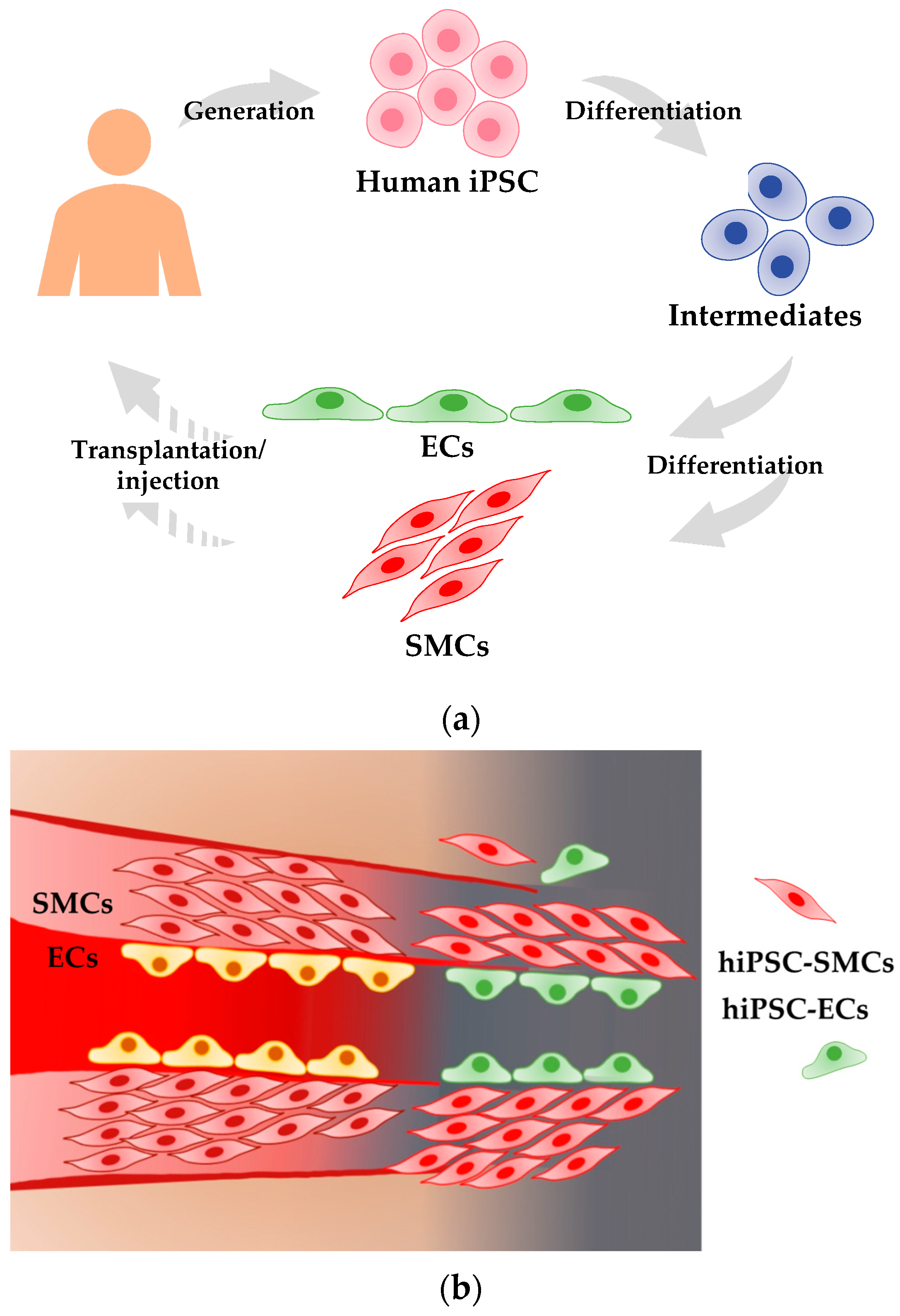
Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions


Abstract
1. Introduction
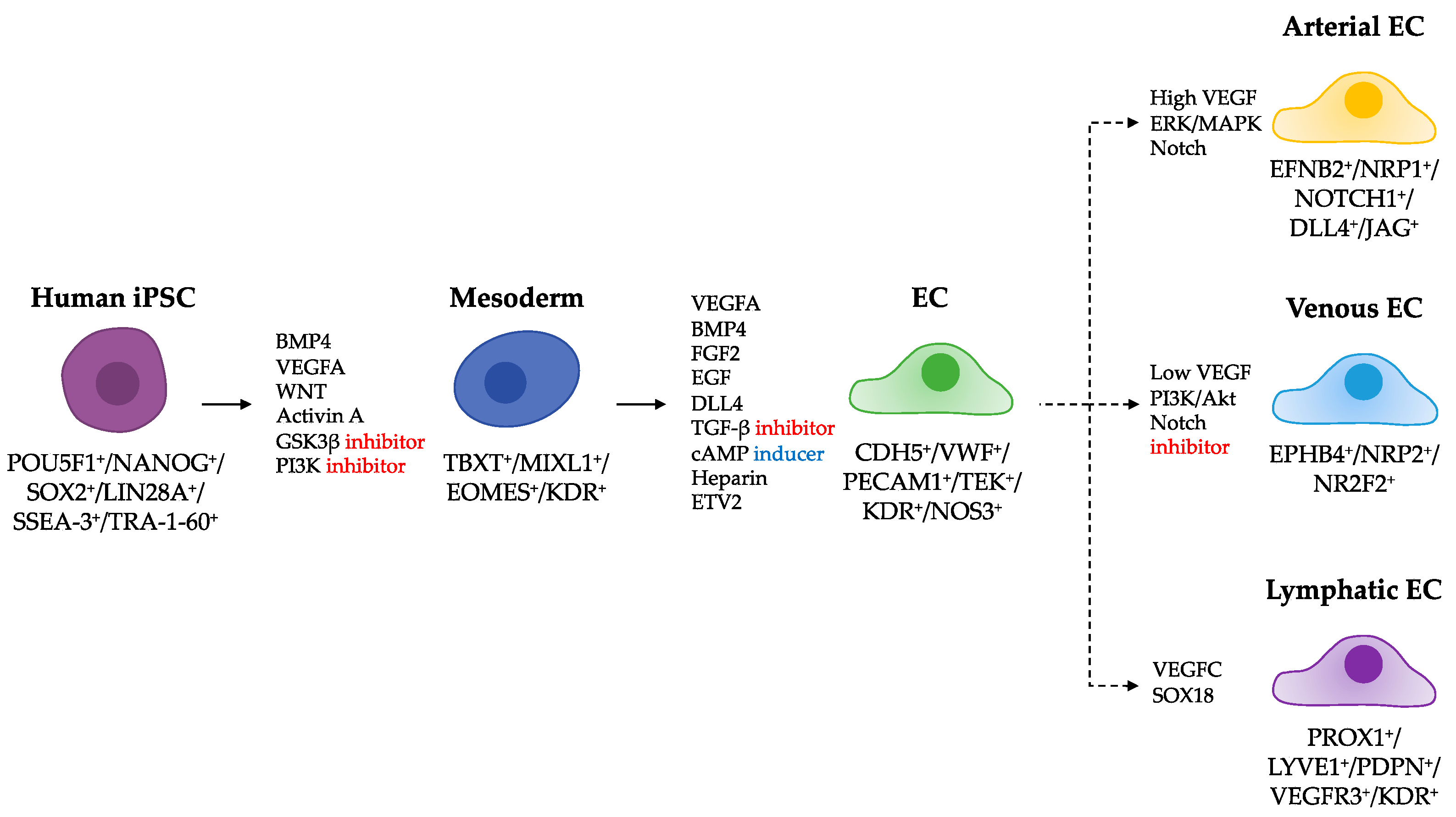
2. Human Induced Pluripotent Stem Cell-Derived Endothelial Cells
2.1. Differentiation of hiPSCs into ECs
2.2. Criteria to Define EC Identity
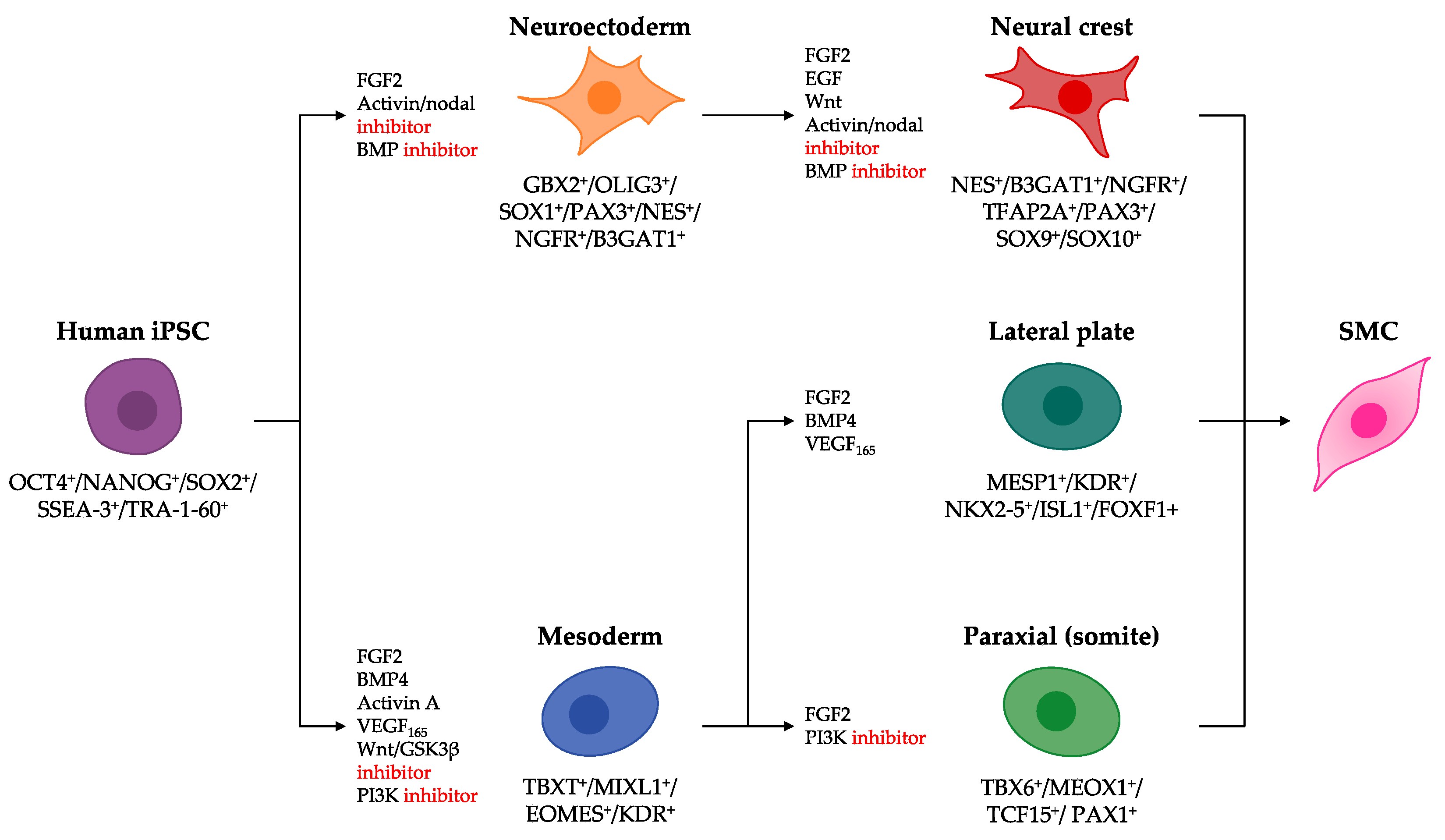
3. Human Induced Pluripotent Stem Cell-Derived Smooth Muscle Cells
3.1. Differentiation of hiPSCs into Lineage-Specific SMC Intermediates
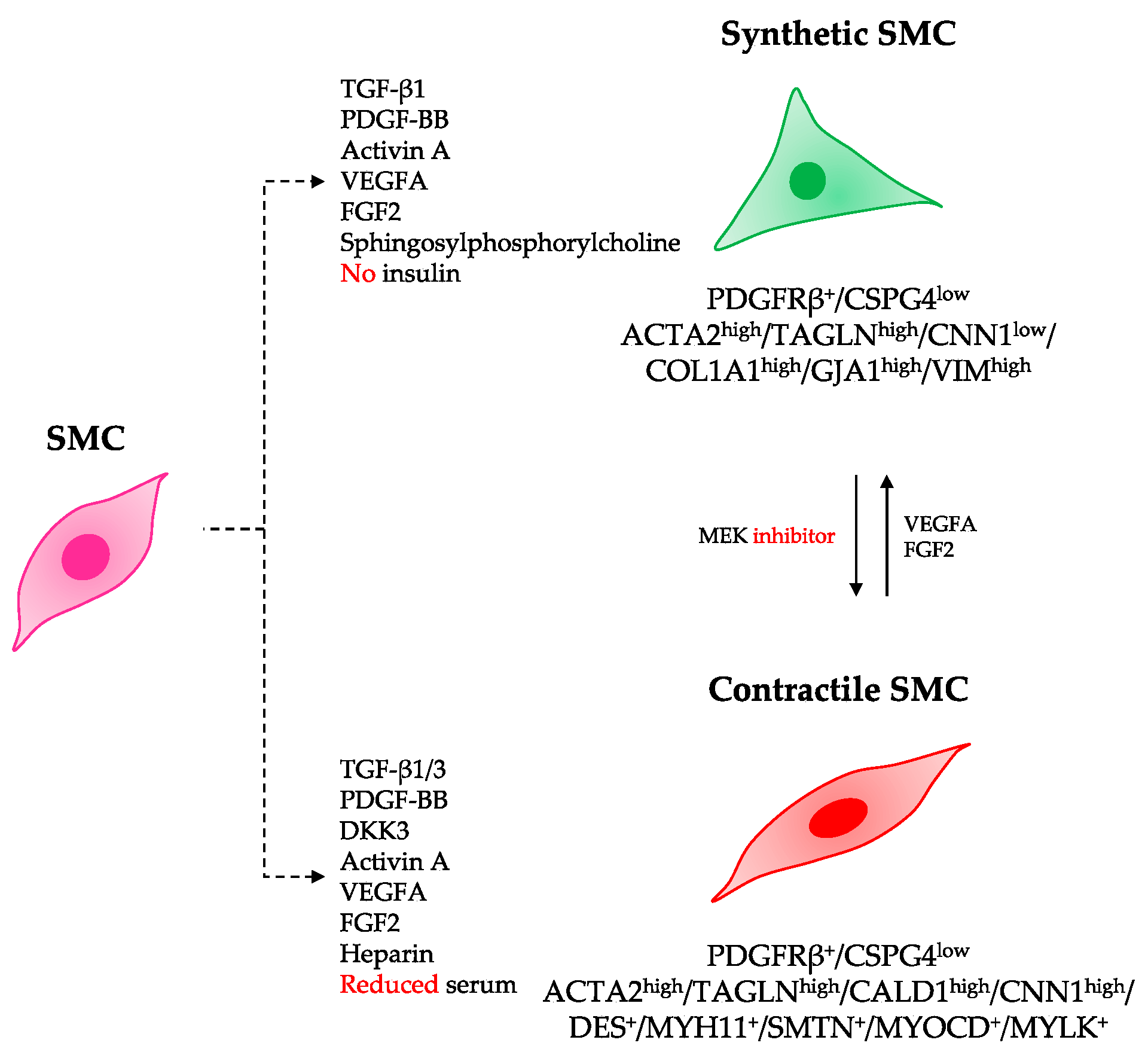
3.2. Differentiation of hiPSCs into Specialized SMC Phenotypes
3.3. Criteria to Define the Contractile SMC Identity
4. Therapeutic Applications Using hiPSC-Derived Vascular Cells
5. Challenges and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcLDL | Acetylated low-density lipoprotein |
| ACTA2 | Actin alpha 2, smooth muscle |
| ANGPT2 | Angiopoietin-2 |
| APLNR | Apelin receptor |
| ATAC-seq | Assay for transposase-accessible chromatin using sequencing |
| B3GAT1 | Beta-1,3-glucuronyltransferase 1 |
| BLI | Bioluminescence imaging |
| BMP4 | Bone morphogenetic protein 4 |
| CALD1 | Caldesmon 1 |
| CB-ECFC | Cord-blood endothelial colony-forming cell |
| CD34 | CD34 molecule |
| CD44 | CD44 molecule (Indian blood group) |
| CDH5 | Cadherin 5 |
| CMC | Chemistry, Manufacturing, and Control |
| cAMP | Cyclic adenosine monophosphate |
| CNN1 | Calponin 1 |
| COL1A1 | Collagen type I alpha 1 chain |
| NR2F2 | Nuclear receptor subfamily 2 group F member 2 |
| CRIP1 and 2 | Cysteine rich protein 1 and 2 |
| DES | Desmin |
| DLL4 | Delta like canonical Notch ligand 4 |
| EB | Embryoid body |
| EC | Endothelial cell |
| EFNB2 | Ephrin B2 |
| EGF | Epidermal growth factor |
| eGFP | Enhanced green fluorescent protein |
| ELISA | Enzyme-linked immunosorbent assay |
| ENG | Endoglin |
| EOMES | Eomesodermin |
| EPHB4 | EphB4 |
| ERG | ETS-related gene |
| FACS | Fluorescence-activated cell sorting |
| FBS | Fetal bovine serum |
| FGF | Fibroblast growth factor |
| FGF2 | Fibroblast growth factor 2 |
| FOXF1 | Forkhead box F1 |
| GBX2 | Gastrulation brain homeobox 2 |
| GJA1 | Gap junction protein alpha 1 |
| GMP | Good manufacturing practice |
| GO term | Gene Ontology term |
| GSK3β | Glycogen synthase kinase 3 beta |
| hBM | Human bone marrow mononuclear cell |
| hESC | Human embryonic stem cell |
| hiPSC | Human induced pluripotent stem cell |
| HUVEC | Human umbilical cord endothelial cell |
| iEnd | induced EC via transfection of fibroblasts with POU5F1 and KLF4; ISL1, ISL LIM homeobox 1 |
| ITGA2 | Integrin subunit alpha 2 |
| KDR | Kinase insert domain receptor |
| KEGG pathway | Database of metabolic pathways from Kyoto Encyclopedia of Genes and Genomes |
| KLF4 | Kruppel like factor 4 |
| LIN28A | Lin-28 homolog A |
| LMOD1 | Leiomodin 1 |
| Lrp5 and 6 | Low density lipoprotein receptor-related protein 5 and 6 |
| MAP2K7 | Mitogen-activated protein kinase 7 |
| MCAM | Melanoma cell adhesion molecule |
| MEOX1 | Mesenchyme homeobox 1 |
| MITCH-PEG | Mixing-Induced Two-Component Hydrogels combined with PolyEthylene Glycol hybrid hydrogel |
| MIXL1 | Mix paired-like homeobox 1 |
| MRTFA and B | Myocardin related transcription factor A and B |
| MYC | MYC proto-oncogene, bHLH transcription factor |
| MYH11 | Myosin heavy chain 11 |
| MYH11b | Myosin, heavy chain 11b, smooth muscle (Danio rerio (zebrafish)) |
| MYLK | Myosin light chain kinase |
| MYOCD | Myocardin |
| NANOG | Nanog homeobox |
| NES | Nestin |
| NGFR | Nerve growth factor receptor |
| NKX2-5 | NK2 homeobox 5 |
| NO | Nitric oxide |
| NOTCH1 | Notch receptor 1 |
| NOS3 | Nitric oxide synthase 3 |
| NRP1 | Neuropilin 1 |
| NT5E | 5′-Nucleotidase ecto |
| OLIG3 | Oligodendrocyte transcription factor 3 |
| PAX1, 3, and 7 | Paired box 1, 3, and 7 |
| PDGFB | Platelet-derived growth factor subunit B |
| PDGFR⍺ and β | Platelet-derived growth factor receptor alpha and beta |
| PECAM1 | Platelet and endothelial cell adhesion molecule 1 |
| PI3K | Phosphatidylinositol-4,5-biphosphate 3-kinase |
| PITX2 | Paired like homeodomain 2 |
| POU5F1 | POU class 5 homeobox 1 |
| RA | Retinoic acid |
| ROCK | Rho-associated protein kinase |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SHIELD | Shear-thinning Hydrogel for Injectable Encapsulation and Long-term Delivery |
| SMC | Smooth muscle cell |
| SMTN | Smoothelin |
| SOX1, 2, 9, and 10 | SRY-box transcription factor 1, 2, 9, and 10 |
| SRF | Serum response factor |
| SYNPO2 | Synaptopodin 2 |
| TAGLN | Transgelin |
| TBX6 and T | T-box transcription factor 6 and T |
| TCF15 | Transcription factor 15 |
| TEK | Tyrosine kinase |
| TFAP2A | Transcription factor AP-2 alpha |
| TGF-β | Transforming growth factor-beta |
| THY1 | Thy-1 cell surface antigen |
| VEGF | Vascular endothelial growth factor |
| VEGFA | Vascular endothelial growth factor A |
| VIM | Vimentin |
| VWF | von Willebrand factor |
| Wnt | Wingless/Integrated |
| Wnt3 | Wingless-type MMTV integration site family, member 3 |
References
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 12 August 2021).
- Lopez, A.D.; Murray, C.C. The global burden of disease, 1990–2020. Nat. Med. 1998, 4, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Schillinger, M.; Sabeti, S.; Loewe, C.; Dick, P.; Amighi, J.; Mlekusch, W.; Schlager, O.; Cejna, M.; Lammer, J.; Minar, E. Balloon angioplasty versus implantation of nitinol stents in the superficial femoral artery. N. Engl. J. Med. 2006, 354, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Warfarin Antiplatelet Vascular Evaluation Trial, I.; Anand, S.; Yusuf, S.; Xie, C.; Pogue, J.; Eikelboom, J.; Budaj, A.; Sussex, B.; Liu, L.; Guzman, R.; et al. Oral anticoagulant and antiplatelet therapy and peripheral arterial disease. N. Engl. J. Med. 2007, 357, 217–227. [Google Scholar] [CrossRef]
- Serruys, P.W.; Morice, M.C.; Kappetein, A.P.; Colombo, A.; Holmes, D.R.; Mack, M.J.; Stahle, E.; Feldman, T.E.; van den Brand, M.; Bass, E.J.; et al. Percutaneous coronary intervention versus coronary-artery bypass grafting for severe coronary artery disease. N. Engl. J. Med. 2009, 360, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, W.S.; Grau-Sepulveda, M.V.; Weiss, J.M.; O’Brien, S.M.; Peterson, E.D.; Kolm, P.; Zhang, Z.; Klein, L.W.; Shaw, R.E.; McKay, C.; et al. Comparative effectiveness of revascularization strategies. N. Engl. J. Med. 2012, 366, 1467–1476. [Google Scholar] [CrossRef]
- Waldo, S.W.; Secemsky, E.A.; O’Brien, C.; Kennedy, K.F.; Pomerantsev, E.; Sundt, T.M., 3rd; McNulty, E.J.; Scirica, B.M.; Yeh, R.W. Surgical ineligibility and mortality among patients with unprotected left main or multivessel coronary artery disease undergoing percutaneous coronary intervention. Circulation 2014, 130, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Isner, J.M.; Asahara, T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J. Clin. Investig. 1999, 103, 1231–1236. [Google Scholar] [CrossRef] [PubMed]
- Losordo, D.W.; Dimmeler, S. Therapeutic angiogenesis and vasculogenesis for ischemic disease: Part II: Cell-based therapies. Circulation 2004, 109, 2692–2697. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. Angiogenesis: Where do we stand now? Circulation 2005, 111, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol 1995, 11, 73–91. [Google Scholar] [CrossRef]
- Risau, W. Differentiation of endothelium. FASEB J. 1995, 9, 926–933. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar] [CrossRef]
- Geudens, I.; Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development 2011, 138, 4569–4583. [Google Scholar] [CrossRef]
- Moraes, F.; Paye, J.; Mac Gabhann, F.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef]
- Losordo, D.W.; Dimmeler, S. Therapeutic angiogenesis and vasculogenesis for ischemic disease. Part I: Angiogenic cytokines. Circulation 2004, 109, 2487–2491. [Google Scholar] [CrossRef]
- Liu, H.; Kennard, S.; Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 2009, 104, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Simons, M. Regulation of vascular integrity. J. Mol. Med. 2009, 87, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Frontini, M.J.; Nong, Z.; Gros, R.; Drangova, M.; O’Neil, C.; Rahman, M.N.; Akawi, O.; Yin, H.; Ellis, C.G.; Pickering, J.G. Fibroblast growth factor 9 delivery during angiogenesis produces durable, vasoresponsive microvessels wrapped by smooth muscle cells. Nat. Biotechnol. 2011, 29, 421–427. [Google Scholar] [CrossRef]
- Buschmann, I.; Schaper, W. Arteriogenesis Versus Angiogenesis: Two Mechanisms of Vessel Growth. News Physiol. Sci. 1999, 14, 121–125. [Google Scholar] [CrossRef]
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W.; Scholz, D. Factors regulating arteriogenesis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.A.; Majka, S.M.; Wang, H.; Pocius, J.; Hartley, C.J.; Majesky, M.W.; Entman, M.L.; Michael, L.H.; Hirschi, K.K.; Goodell, M.A. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J. Clin. Investig. 2001, 107, 1395–1402. [Google Scholar] [CrossRef]
- Kawamoto, A.; Gwon, H.C.; Iwaguro, H.; Yamaguchi, J.I.; Uchida, S.; Masuda, H.; Silver, M.; Ma, H.; Kearney, M.; Isner, J.M.; et al. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation 2001, 103, 634–637. [Google Scholar] [CrossRef]
- Nagaya, N.; Fujii, T.; Iwase, T.; Ohgushi, H.; Itoh, T.; Uematsu, M.; Yamagishi, M.; Mori, H.; Kangawa, K.; Kitamura, S. Intravenous administration of mesenchymal stem cells improves cardiac function in rats with acute myocardial infarction through angiogenesis and myogenesis. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2670–H2676. [Google Scholar] [CrossRef]
- Ma, N.; Stamm, C.; Kaminski, A.; Li, W.; Kleine, H.D.; Muller-Hilke, B.; Zhang, L.; Ladilov, Y.; Egger, D.; Steinhoff, G. Human cord blood cells induce angiogenesis following myocardial infarction in NOD/scid-mice. Cardiovasc. Res. 2005, 66, 45–54. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Li, T.S.; Nishida, M.; Ito, H.; Matsuzaki, M.; Kasaoka, S.; Hamano, K. Autologous bone marrow cell implantation as therapeutic angiogenesis for ischemic hindlimb in diabetic rat model. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H66–H70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iwase, T.; Nagaya, N.; Fujii, T.; Itoh, T.; Murakami, S.; Matsumoto, T.; Kangawa, K.; Kitamura, S. Comparison of angiogenic potency between mesenchymal stem cells and mononuclear cells in a rat model of hindlimb ischemia. Cardiovasc. Res. 2005, 66, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Strauer, B.E.; Brehm, M.; Zeus, T.; Kostering, M.; Hernandez, A.; Sorg, R.V.; Kogler, G.; Wernet, P. Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation 2002, 106, 1913–1918. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Dubois, C.; Bogaert, J.; Theunissen, K.; Deroose, C.; Desmet, W.; Kalantzi, M.; Herbots, L.; Sinnaeve, P.; Dens, J.; et al. Autologous bone marrow-derived stem-cell transfer in patients with ST-segment elevation myocardial infarction: Double-blind, randomised controlled trial. Lancet 2006, 367, 113–121. [Google Scholar] [CrossRef]
- Lunde, K.; Solheim, S.; Aakhus, S.; Arnesen, H.; Abdelnoor, M.; Egeland, T.; Endresen, K.; Ilebekk, A.; Mangschau, A.; Fjeld, J.G.; et al. Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. N. Engl. J. Med. 2006, 355, 1199–1209. [Google Scholar] [CrossRef]
- Meyer, G.P.; Wollert, K.C.; Lotz, J.; Pirr, J.; Rager, U.; Lippolt, P.; Hahn, A.; Fichtner, S.; Schaefer, A.; Arseniev, L.; et al. Intracoronary bone marrow cell transfer after myocardial infarction: 5-year follow-up from the randomized-controlled BOOST trial. Eur. Heart J. 2009, 30, 2978–2984. [Google Scholar] [CrossRef] [PubMed]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef]
- Cho, H.J.; Lee, N.; Lee, J.Y.; Choi, Y.J.; Ii, M.; Wecker, A.; Jeong, J.O.; Curry, C.; Qin, G.; Yoon, Y.S. Role of host tissues for sustained humoral effects after endothelial progenitor cell transplantation into the ischemic heart. J. Exp. Med. 2007, 204, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yoon, Y.S. Revisiting cardiovascular regeneration with bone marrow-derived angiogenic and vasculogenic cells. Br. J. Pharmacol. 2013, 169, 290–303. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Reubinoff, B.E.; Pera, M.F.; Fong, C.Y.; Trounson, A.; Bongso, A. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat. Biotechnol. 2000, 18, 399–404. [Google Scholar] [CrossRef]
- Amit, M.; Carpenter, M.K.; Inokuma, M.S.; Chiu, C.P.; Harris, C.P.; Waknitz, M.A.; Itskovitz-Eldor, J.; Thomson, J.A. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev. Biol. 2000, 227, 271–278. [Google Scholar] [CrossRef]
- Yamashita, J.; Itoh, H.; Hirashima, M.; Ogawa, M.; Nishikawa, S.; Yurugi, T.; Naito, M.; Nakao, K.; Nishikawa, S. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 2000, 408, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Levenberg, S.; Golub, J.S.; Amit, M.; Itskovitz-Eldor, J.; Langer, R. Endothelial cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 4391–4396. [Google Scholar] [CrossRef]
- Gerecht-Nir, S.; Ziskind, A.; Cohen, S.; Itskovitz-Eldor, J. Human embryonic stem cells as an in vitro model for human vascular development and the induction of vascular differentiation. Lab. Investig. 2003, 83, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Au, P.; Chen, T.; Shao, Y.; Daheron, L.M.; Bai, H.; Arzigian, M.; Fukumura, D.; Jain, R.K.; Scadden, D.T. Endothelial cells derived from human embryonic stem cells form durable blood vessels in vivo. Nat. Biotechnol. 2007, 25, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Levenberg, S.; Ferreira, L.S.; Chen-Konak, L.; Kraehenbuehl, T.P.; Langer, R. Isolation, differentiation and characterization of vascular cells derived from human embryonic stem cells. Nat. Protoc. 2010, 5, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, J.C.; Sheikh, A.Y.; Kraft, D.; Cao, F.; Xie, X.; Patel, M.; Gambhir, S.S.; Robbins, R.C.; Cooke, J.P.; et al. Differentiation, survival, and function of embryonic stem cell derived endothelial cells for ischemic heart disease. Circulation 2007, 116, I46–I54. [Google Scholar] [CrossRef]
- Li, Z.; Wilson, K.D.; Smith, B.; Kraft, D.L.; Jia, F.; Huang, M.; Xie, X.; Robbins, R.C.; Gambhir, S.S.; Weissman, I.L.; et al. Functional and transcriptional characterization of human embryonic stem cell-derived endothelial cells for treatment of myocardial infarction. PLoS ONE 2009, 4, e8443. [Google Scholar] [CrossRef]
- Cho, S.W.; Moon, S.H.; Lee, S.H.; Kang, S.W.; Kim, J.; Lim, J.M.; Kim, H.S.; Kim, B.S.; Chung, H.M. Improvement of postnatal neovascularization by human embryonic stem cell derived endothelial-like cell transplantation in a mouse model of hindlimb ischemia. Circulation 2007, 116, 2409–2419. [Google Scholar] [CrossRef]
- Huang, N.F.; Niiyama, H.; Peter, C.; De, A.; Natkunam, Y.; Fleissner, F.; Li, Z.; Rollins, M.D.; Wu, J.C.; Gambhir, S.S.; et al. Embryonic stem cell-derived endothelial cells engraft into the ischemic hindlimb and restore perfusion. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 984–991. [Google Scholar] [CrossRef]
- Moon, S.H.; Kim, J.S.; Park, S.J.; Lee, H.J.; Do, J.T.; Chung, H.M. A system for treating ischemic disease using human embryonic stem cell-derived endothelial cells without direct incorporation. Biomaterials 2011, 32, 6445–6455. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef]
- Haase, A.; Olmer, R.; Schwanke, K.; Wunderlich, S.; Merkert, S.; Hess, C.; Zweigerdt, R.; Gruh, I.; Meyer, J.; Wagner, S.; et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 2009, 5, 434–441. [Google Scholar] [CrossRef]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Zhong, X.; Li, N.; Liang, S.; Huang, Q.; Coukos, G.; Zhang, L. Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J. Biol. Chem. 2010, 285, 41961–41971. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Rossi, D.J. Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat. Protoc. 2013, 8, 568–582. [Google Scholar] [CrossRef]
- Han, J.W.; Yoon, Y.S. Induced pluripotent stem cells: Emerging techniques for nuclear reprogramming. Antioxid. Redox Signal. 2011, 15, 1799–1820. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Taura, D.; Sone, M.; Homma, K.; Oyamada, N.; Takahashi, K.; Tamura, N.; Yamanaka, S.; Nakao, K. Induction and isolation of vascular cells from human induced pluripotent stem cells—Brief report. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1100–1103. [Google Scholar] [CrossRef]
- Rufaihah, A.J.; Huang, N.F.; Jame, S.; Lee, J.C.; Nguyen, H.N.; Byers, B.; De, A.; Okogbaa, J.; Rollins, M.; Reijo-Pera, R.; et al. Endothelial cells derived from human iPSCS increase capillary density and improve perfusion in a mouse model of peripheral arterial disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e72–e79. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Gil, C.H.; Yoder, M.C. Differentiation, Evaluation, and Application of Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2014–2025. [Google Scholar] [CrossRef]
- Xu, M.; He, J.; Zhang, C.; Xu, J.; Wang, Y. Strategies for derivation of endothelial lineages from human stem cells. Stem Cell Res. Ther. 2019, 10, 200. [Google Scholar] [CrossRef]
- Lee, S.J.; Sohn, Y.D.; Andukuri, A.; Kim, S.; Byun, J.; Han, J.W.; Park, I.H.; Jun, H.W.; Yoon, Y.S. Enhanced Therapeutic and Long-Term Dynamic Vascularization Effects of Human Pluripotent Stem Cell-Derived Endothelial Cells Encapsulated in a Nanomatrix Gel. Circulation 2017, 136, 1939–1954. [Google Scholar] [CrossRef]
- Belair, D.G.; Whisler, J.A.; Valdez, J.; Velazquez, J.; Molenda, J.A.; Vickerman, V.; Lewis, R.; Daigh, C.; Hansen, T.D.; Mann, D.A.; et al. Human vascular tissue models formed from human induced pluripotent stem cell derived endothelial cells. Stem Cell Rev. Rep. 2015, 11, 511–525. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, K.H.; Yoon, Y.S. Generation of Human Pluripotent Stem Cell-derived Endothelial Cells and Their Therapeutic Utility. Curr. Cardiol. Rep. 2018, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.; Cortez-Toledo, E.; Magner, N.L.; Beegle, J.R.; Coleal-Bergum, D.P.; Hao, D.; Wang, A.; Nolta, J.A.; Zhou, P. Highly Efficient Differentiation of Endothelial Cells from Pluripotent Stem Cells Requires the MAPK and the PI3K Pathways. Stem Cells 2017, 35, 909–919. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Goldie, L.C.; Hirschi, K.K. Regulation of endothelial cell differentiation and specification. Circ. Res. 2013, 112, 1272–1287. [Google Scholar] [CrossRef]
- Kennedy, C.C.; Brown, E.E.; Abutaleb, N.O.; Truskey, G.A. Development and Application of Endothelial Cells Derived From Pluripotent Stem Cells in Microphysiological Systems Models. Front. Cardiovasc. Med. 2021, 8, 625016. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.E., 3rd; Kelley, R.W.; Patterson, C. Mechanisms of endothelial differentiation in embryonic vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; McGrath, K.E.; Kingsley, P.D. Initiation of hematopoiesis and vasculogenesis in murine yolk sac explants. Blood 1995, 86, 156–163. [Google Scholar] [CrossRef]
- Liu, P.; Wakamiya, M.; Shea, M.J.; Albrecht, U.; Behringer, R.R.; Bradley, A. Requirement for Wnt3 in vertebrate axis formation. Nat. Genet. 1999, 22, 361–365. [Google Scholar] [CrossRef]
- Kelly, O.G.; Pinson, K.I.; Skarnes, W.C. The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development 2004, 131, 2803–2815. [Google Scholar] [CrossRef]
- Huelsken, J.; Vogel, R.; Brinkmann, V.; Erdmann, B.; Birchmeier, C.; Birchmeier, W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 2000, 148, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Gill, J.G.; Kyba, M.; Murphy, T.L.; Murphy, K.M. Canonical Wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development 2006, 133, 3787–3796. [Google Scholar] [CrossRef]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Candia, A.F.; Watabe, T.; Hawley, S.H.; Onichtchouk, D.; Zhang, Y.; Derynck, R.; Niehrs, C.; Cho, K.W. Cellular interpretation of multiple TGF-beta signals: Intracellular antagonism between activin/BVg1 and BMP-2/4 signaling mediated by Smads. Development 1997, 124, 4467–4480. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, N.; Lu, C.; Guzman-Ayala, M.; Pescatore, L.; Mesnard, D.; Bischofberger, M.; Naef, F.; Robertson, E.J.; Constam, D.B. The nodal precursor acting via activin receptors induces mesoderm by maintaining a source of its convertases and BMP4. Dev. Cell 2006, 11, 313–323. [Google Scholar] [CrossRef]
- Rossant, J.; Tam, P.P. Emerging asymmetry and embryonic patterning in early mouse development. Dev. Cell 2004, 7, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Warmflash, A.; Sorre, B.; Etoc, F.; Siggia, E.D.; Brivanlou, A.H. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat. Methods 2014, 11, 847–854. [Google Scholar] [CrossRef]
- Martyn, I.; Kanno, T.Y.; Ruzo, A.; Siggia, E.D.; Brivanlou, A.H. Self-organization of a human organizer by combined Wnt and Nodal signalling. Nature 2018, 558, 132–135. [Google Scholar] [CrossRef]
- Kreuser, U.; Buchert, J.; Haase, A.; Richter, W.; Diederichs, S. Initial WNT/beta-Catenin Activation Enhanced Mesoderm Commitment, Extracellular Matrix Expression, Cell Aggregation and Cartilage Tissue Yield From Induced Pluripotent Stem Cells. Front. Cell Dev. Biol 2020, 8, 581331. [Google Scholar] [CrossRef]
- Amaya, E.; Musci, T.J.; Kirschner, M.W. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell 1991, 66, 257–270. [Google Scholar] [CrossRef]
- Amaya, E.; Stein, P.A.; Musci, T.J.; Kirschner, M.W. FGF signalling in the early specification of mesoderm in Xenopus. Development 1993, 118, 477–487. [Google Scholar] [CrossRef]
- Griffin, K.; Patient, R.; Holder, N. Analysis of FGF function in normal and no tail zebrafish embryos reveals separate mechanisms for formation of the trunk and the tail. Development 1995, 121, 2983–2994. [Google Scholar] [CrossRef]
- Fletcher, R.B.; Harland, R.M. The role of FGF signaling in the establishment and maintenance of mesodermal gene expression in Xenopus. Dev. Dyn. 2008, 237, 1243–1254. [Google Scholar] [CrossRef]
- Carballada, R.; Yasuo, H.; Lemaire, P. Phosphatidylinositol-3 kinase acts in parallel to the ERK MAP kinase in the FGF pathway during Xenopus mesoderm induction. Development 2001, 128, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.P.; Harpal, K.; Henkemeyer, M.; Rossant, J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994, 8, 3032–3044. [Google Scholar] [CrossRef]
- Xu, R.H.; Peck, R.M.; Li, D.S.; Feng, X.; Ludwig, T.; Thomson, J.A. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods 2005, 2, 185–190. [Google Scholar] [CrossRef]
- Yu, P.; Pan, G.; Yu, J.; Thomson, J.A. FGF2 sustains NANOG and switches the outcome of BMP4-induced human embryonic stem cell differentiation. Cell Stem Cell 2011, 8, 326–334. [Google Scholar] [CrossRef]
- Sudheer, S.; Bhushan, R.; Fauler, B.; Lehrach, H.; Adjaye, J. FGF inhibition directs BMP4-mediated differentiation of human embryonic stem cells to syncytiotrophoblast. Stem Cells Dev. 2012, 21, 2987–3000. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.M.; Reynolds, D.; Cliff, T.; Ohtsuka, S.; Mattheyses, A.L.; Sun, Y.; Menendez, L.; Kulik, M.; Dalton, S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 2012, 10, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Tsuneyoshi, N.; Nakatsuji, N.; Suemori, H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development 2008, 135, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Drabsch, Y.; Freund, C.; Petrus-Reurer, S.; van den Hil, F.E.; Muenthaisong, S.; Dijke, P.T.; Mummery, C.L. Functionality of endothelial cells and pericytes from human pluripotent stem cells demonstrated in cultured vascular plexus and zebrafish xenografts. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 177–186. [Google Scholar] [CrossRef]
- Lian, X.; Bao, X.; Al-Ahmad, A.; Liu, J.; Wu, Y.; Dong, W.; Dunn, K.K.; Shusta, E.V.; Palecek, S.P. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Rep. 2014, 3, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qi, J.; Xu, X.; Zeisberg, M.; Guan, K.; Zeisberg, E.M. Differentiation of functional endothelial cells from human induced pluripotent stem cells: A novel, highly efficient and cost effective method. Differentiation 2016, 92, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lin, R.Z.; Hong, X.; Ng, A.H.; Lee, C.N.; Neumeyer, J.; Wang, G.; Wang, X.; Ma, M.; Pu, W.T.; et al. Robust differentiation of human pluripotent stem cells into endothelial cells via temporal modulation of ETV2 with modified mRNA. Sci. Adv. 2020, 6, eaba7606. [Google Scholar] [CrossRef]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef]
- Goldman, O.; Feraud, O.; Boyer-Di Ponio, J.; Driancourt, C.; Clay, D.; Le Bousse-Kerdiles, M.C.; Bennaceur-Griscelli, A.; Uzan, G. A boost of BMP4 accelerates the commitment of human embryonic stem cells to the endothelial lineage. Stem Cells 2009, 27, 1750–1759. [Google Scholar] [CrossRef]
- Choi, K.D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ii, M.; Kamei, N.; Alev, C.; Kwon, S.M.; Kawamoto, A.; Akimaru, H.; Masuda, H.; Sawa, Y.; Asahara, T. CD34+ cells represent highly functional endothelial progenitor cells in murine bone marrow. PLoS ONE 2011, 6, e20219. [Google Scholar] [CrossRef] [PubMed]
- Prasain, N.; Lee, M.R.; Vemula, S.; Meador, J.L.; Yoshimoto, M.; Ferkowicz, M.J.; Fett, A.; Gupta, M.; Rapp, B.M.; Saadatzadeh, M.R.; et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 2014, 32, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, N.F.; Zou, J.; Laurent, T.J.; Lee, J.C.; Okogbaa, J.; Cooke, J.P.; Ding, S. Conversion of human fibroblasts to functional endothelial cells by defined factors. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1366–1375. [Google Scholar] [CrossRef]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Nourse, M.B.; Halpin, D.E.; Scatena, M.; Mortisen, D.J.; Tulloch, N.L.; Hauch, K.D.; Torok-Storb, B.; Ratner, B.D.; Pabon, L.; Murry, C.E. VEGF induces differentiation of functional endothelium from human embryonic stem cells: Implications for tissue engineering. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Huylebroeck, D.; Verschueren, K.; Guo, Q.; Matzuk, M.M.; Zwijsen, A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development 1999, 126, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, C.; Xu, X.; Deng, C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3667–3672. [Google Scholar] [CrossRef]
- Ribatti, D.; Urbinati, C.; Nico, B.; Rusnati, M.; Roncali, L.; Presta, M. Endogenous basic fibroblast growth factor is implicated in the vascularization of the chick embryo chorioallantoic membrane. Dev. Biol. 1995, 170, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef]
- Correia, A.C.; Moonen, J.R.; Brinker, M.G.; Krenning, G. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-beta signaling. J. Cell Sci. 2016, 129, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: Implications for modulating arteriogenesis and angiogenesis. Mol. Cell Biol. 2003, 23, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiang, M.; Liu, Y.; Sun, N.; Lu, M.; Shi, Y.; Wang, X.; Meng, D.; Chen, S.; Qin, J. Human induced pluripotent stem cells derived endothelial cells mimicking vascular inflammatory response under flow. Biomicrofluidics 2016, 10, 014106. [Google Scholar] [CrossRef] [PubMed]
- Ikuno, T.; Masumoto, H.; Yamamizu, K.; Yoshioka, M.; Minakata, K.; Ikeda, T.; Sakata, R.; Yamashita, J.K. Efficient and robust differentiation of endothelial cells from human induced pluripotent stem cells via lineage control with VEGF and cyclic AMP. PLoS ONE 2017, 12, e0173271. [Google Scholar] [CrossRef]
- Paik, D.T.; Tian, L.; Lee, J.; Sayed, N.; Chen, I.Y.; Rhee, S.; Rhee, J.W.; Kim, Y.; Wirka, R.C.; Buikema, J.W.; et al. Large-Scale Single-Cell RNA-Seq Reveals Molecular Signatures of Heterogeneous Populations of Human Induced Pluripotent Stem Cell-Derived Endothelial Cells. Circ. Res. 2018, 123, 443–450. [Google Scholar] [CrossRef]
- Sumanas, S.; Jorniak, T.; Lin, S. Identification of novel vascular endothelial-specific genes by the microarray analysis of the zebrafish cloche mutants. Blood 2005, 106, 534–541. [Google Scholar] [CrossRef] [PubMed]
- De Val, S.; Black, B.L. Transcriptional control of endothelial cell development. Dev. Cell 2009, 16, 180–195. [Google Scholar] [CrossRef] [PubMed]
- De Val, S.; Chi, N.C.; Meadows, S.M.; Minovitsky, S.; Anderson, J.P.; Harris, I.S.; Ehlers, M.L.; Agarwal, P.; Visel, A.; Xu, S.M.; et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell 2008, 135, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.; Brok-Volchanskaya, V.; Kumar, A.; Liu, P.; Lee, J.H.; Tong, L.; Vodyanik, M.; Swanson, S.; Stewart, R.; Kyba, M.; et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat. Commun. 2014, 5, 4372. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, A.G.; Veldman, M.B.; Lin, S. ETV2 expression increases the efficiency of primitive endothelial cell derivation from human embryonic stem cells. Cell Regen. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Suknuntha, K.; Tao, L.; Brok-Volchanskaya, V.; D’Souza, S.S.; Kumar, A.; Slukvin, I. Optimization of Synthetic mRNA for Highly Efficient Translation and its Application in the Generation of Endothelial and Hematopoietic Cells from Human and Primate Pluripotent Stem Cells. Stem Cell Rev. Rep. 2018, 14, 525–534. [Google Scholar] [CrossRef]
- Lee, S.; Park, C.; Han, J.W.; Kim, J.Y.; Cho, K.; Kim, E.J.; Kim, S.; Lee, S.J.; Oh, S.Y.; Tanaka, Y.; et al. Direct Reprogramming of Human Dermal Fibroblasts Into Endothelial Cells Using ER71/ETV2. Circ. Res. 2017, 120, 848–861. [Google Scholar] [CrossRef]
- Kume, T. Specification of arterial, venous, and lymphatic endothelial cells during embryonic development. Histol. Histopathol. 2010, 25, 637–646. [Google Scholar] [CrossRef]
- Swift, M.R.; Weinstein, B.M. Arterial-venous specification during development. Circ. Res. 2009, 104, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Rufaihah, A.J.; Huang, N.F.; Kim, J.; Herold, J.; Volz, K.S.; Park, T.S.; Lee, J.C.; Zambidis, E.T.; Reijo-Pera, R.; Cooke, J.P. Human induced pluripotent stem cell-derived endothelial cells exhibit functional heterogeneity. Am. J. Transl. Res. 2013, 5, 21–35. [Google Scholar]
- Arora, S.; Yim, E.K.F.; Toh, Y.C. Environmental Specification of Pluripotent Stem Cell Derived Endothelial Cells Toward Arterial and Venous Subtypes. Front. Bioeng. Biotechnol. 2019, 7, 143. [Google Scholar] [CrossRef]
- Wang, H.U.; Chen, Z.F.; Anderson, D.J. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef]
- Corada, M.; Morini, M.F.; Dejana, E. Signaling pathways in the specification of arteries and veins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef]
- Simons, M.; Eichmann, A. Molecular controls of arterial morphogenesis. Circ. Res. 2015, 116, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.; Adams, R.H. Regulation of vascular morphogenesis by Notch signaling. Genes Dev. 2007, 21, 2511–2524. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.; Praca, C.; Pitrez, P.R.; Gouveia, P.J.; Aranguren, X.L.; Ricotti, L.; Ferreira, L.S. Functional characterization of iPSC-derived arterial- and venous-like endothelial cells. Sci. Rep. 2019, 9, 3826. [Google Scholar] [CrossRef]
- Zhang, J.; Chu, L.F.; Hou, Z.; Schwartz, M.P.; Hacker, T.; Vickerman, V.; Swanson, S.; Leng, N.; Nguyen, B.K.; Elwell, A.; et al. Functional characterization of human pluripotent stem cell-derived arterial endothelial cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6072–E6078. [Google Scholar] [CrossRef]
- Haudenschild, C.C. Morphology of vascular endothelial cells in culture. In Biology of Endothelial Cells; Jaffe, E.A., Ed.; Springer: Boston, MA, USA, 1984; pp. 129–140. [Google Scholar]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef]
- Adams, W.J.; Zhang, Y.; Cloutier, J.; Kuchimanchi, P.; Newton, G.; Sehrawat, S.; Aird, W.C.; Mayadas, T.N.; Luscinskas, F.W.; Garcia-Cardena, G. Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 105–113. [Google Scholar] [CrossRef]
- White, M.P.; Rufaihah, A.J.; Liu, L.; Ghebremariam, Y.T.; Ivey, K.N.; Cooke, J.P.; Srivastava, D. Limited gene expression variation in human embryonic stem cell and induced pluripotent stem cell-derived endothelial cells. Stem Cells 2013, 31, 92–103. [Google Scholar] [CrossRef]
- Patel, J.; Seppanen, E.J.; Rodero, M.P.; Wong, H.Y.; Donovan, P.; Neufeld, Z.; Fisk, N.M.; Francois, M.; Khosrotehrani, K. Functional Definition of Progenitors Versus Mature Endothelial Cells Reveals Key SoxF-Dependent Differentiation Process. Circulation 2017, 135, 786–805. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Kojima, H.; Sakurai, K.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Akaike, T.; Maeda, H.; Nagano, T. Development of a fluorescent indicator for the bioimaging of nitric oxide. Biol. Pharm. Bull. 1997, 20, 1229–1232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kojima, H.; Nakatsubo, N.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Detection and imaging of nitric oxide with novel fluorescent indicators: Diaminofluoresceins. Anal. Chem. 1998, 70, 2446–2453. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Hirotani, M.; Nakatsubo, N.; Kikuchi, K.; Urano, Y.; Higuchi, T.; Hirata, Y.; Nagano, T. Bioimaging of nitric oxide with fluorescent indicators based on the rhodamine chromophore. Anal. Chem. 2001, 73, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- DeCicco-Skinner, K.L.; Henry, G.H.; Cataisson, C.; Tabib, T.; Gwilliam, J.C.; Watson, N.J.; Bullwinkle, E.M.; Falkenburg, L.; O’Neill, R.C.; Morin, A.; et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp. 2014, 91, e51312. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Song, S.H.; Kim, K.L.; Yi, J.Y.; Shin, G.H.; Kim, J.Y.; Kim, J.; Han, Y.M.; Lee, S.H.; Lee, S.H.; et al. Functional recapitulation of smooth muscle cells via induced pluripotent stem cells from human aortic smooth muscle cells. Circ. Res. 2010, 106, 120–128. [Google Scholar] [CrossRef]
- Ge, X.; Ren, Y.; Bartulos, O.; Lee, M.Y.; Yue, Z.; Kim, K.Y.; Li, W.; Amos, P.J.; Bozkulak, E.C.; Iyer, A.; et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 2012, 126, 1695–1704. [Google Scholar] [CrossRef]
- Kinnear, C.; Chang, W.Y.; Khattak, S.; Hinek, A.; Thompson, T.; de Carvalho Rodrigues, D.; Kennedy, K.; Mahmut, N.; Pasceri, P.; Stanford, W.L.; et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, J.; Jiao, J.; Liu, Z.; Zhou, Z.; Zhao, C.; Chang, L.J.; Chen, Y.E.; Ma, P.X.; Yang, B. Engineering vascular tissue with functional smooth muscle cells derived from human iPS cells and nanofibrous scaffolds. Biomaterials 2014, 35, 8960–8969. [Google Scholar] [CrossRef]
- Biel, N.M.; Santostefano, K.E.; DiVita, B.B.; El Rouby, N.; Carrasquilla, S.D.; Simmons, C.; Nakanishi, M.; Cooper-DeHoff, R.M.; Johnson, J.A.; Terada, N. Vascular Smooth Muscle Cells From Hypertensive Patient-Derived Induced Pluripotent Stem Cells to Advance Hypertension Pharmacogenomics. Stem Cells Transl. Med. 2015, 4, 1380–1390. [Google Scholar] [CrossRef]
- Majesky, M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef]
- Sinha, S.; Iyer, D.; Granata, A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell Mol. Life Sci. 2014, 71, 2271–2288. [Google Scholar] [CrossRef]
- Topouzis, S.; Majesky, M.W. Smooth muscle lineage diversity in the chick embryo. Two types of aortic smooth muscle cell differ in growth and receptor-mediated transcriptional responses to transforming growth factor-beta. Dev. Biol. 1996, 178, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Gadson, P.F., Jr.; Dalton, M.L.; Patterson, E.; Svoboda, D.D.; Hutchinson, L.; Schram, D.; Rosenquist, T.H. Differential response of mesoderm- and neural crest-derived smooth muscle to TGF-beta1: Regulation of c-myb and alpha1 (I) procollagen genes. Exp. Cell Res. 1997, 230, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, M.; Sesti, C.; Washington, I.M.; Du, L.; Dronadula, N.; Chin, M.T.; Stolz, D.B.; Davis, E.C.; Dichek, D.A. Transforming growth factor-beta signaling in myogenic cells regulates vascular morphogenesis, differentiation, and matrix synthesis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Kengaku, M.; Okamoto, H. Basic fibroblast growth factor induces differentiation of neural tube and neural crest lineages of cultured ectoderm cells from Xenopus gastrula. Development 1993, 119, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Launay, C.; Fromentoux, V.; Shi, D.L.; Boucaut, J.C. A truncated FGF receptor blocks neural induction by endogenous Xenopus inducers. Development 1996, 122, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.; Guerrero, N.; Martinez, C. Role of FGF and noggin in neural crest induction. Dev. Biol 1997, 189, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liem, K.F., Jr.; Tremml, G.; Roelink, H.; Jessell, T.M. Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell 1995, 82, 969–979. [Google Scholar] [CrossRef]
- Kanzler, B.; Foreman, R.K.; Labosky, P.A.; Mallo, M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development 2000, 127, 1095–1104. [Google Scholar] [CrossRef]
- Liu, A.; Niswander, L.A. Bone morphogenetic protein signalling and vertebrate nervous system development. Nat. Rev. Neurosci. 2005, 6, 945–954. [Google Scholar] [CrossRef]
- Saint-Jeannet, J.P.; He, X.; Varmus, H.E.; Dawid, I.B. Regulation of dorsal fate in the neuraxis by Wnt-1 and Wnt-3a. Proc. Natl. Acad. Sci. USA 1997, 94, 13713–13718. [Google Scholar] [CrossRef]
- Leung, A.W.; Murdoch, B.; Salem, A.F.; Prasad, M.S.; Gomez, G.A.; Garcia-Castro, M.I. WNT/beta-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development 2016, 143, 398–410. [Google Scholar] [CrossRef]
- Shah, N.M.; Groves, A.K.; Anderson, D.J. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell 1996, 85, 331–343. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Morita, T. Role of retinoic acid in mouse neural crest cell development in vitro. Dev. Dyn. 1995, 204, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Schlett, K.; Madarasz, E. Retinoic acid induced neural differentiation in a neuroectodermal cell line immortalized by p53 deficiency. J. Neurosci. Res. 1997, 47, 405–415. [Google Scholar] [CrossRef]
- Hemmati-Brivanlou, A.; Melton, D.A. Inhibition of activin receptor signaling promotes neuralization in Xenopus. Cell 1994, 77, 273–281. [Google Scholar] [CrossRef]
- Casellas, R.; Brivanlou, A.H. Xenopus Smad7 inhibits both the activin and BMP pathways and acts as a neural inducer. Dev. Biol. 1998, 198, 1–12. [Google Scholar] [CrossRef][Green Version]
- Villanueva, S.; Glavic, A.; Ruiz, P.; Mayor, R. Posteriorization by FGF, Wnt, and retinoic acid is required for neural crest induction. Dev. Biol. 2002, 241, 289–301. [Google Scholar] [CrossRef]
- Wang, A.; Tang, Z.; Li, X.; Jiang, Y.; Tsou, D.A.; Li, S. Derivation of smooth muscle cells with neural crest origin from human induced pluripotent stem cells. Cells Tissues Organs 2012, 195, 5–14. [Google Scholar] [CrossRef]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef]
- Cheung, C.; Bernardo, A.S.; Pedersen, R.A.; Sinha, S. Directed differentiation of embryonic origin-specific vascular smooth muscle subtypes from human pluripotent stem cells. Nat. Protoc. 2014, 9, 929–938. [Google Scholar] [CrossRef]
- Cheung, C.; Goh, Y.T.; Zhang, J.; Wu, C.; Guccione, E. Modeling cerebrovascular pathophysiology in amyloid-beta metabolism using neural-crest-derived smooth muscle cells. Cell Rep. 2014, 9, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Vallier, L.; Touboul, T.; Chng, Z.; Brimpari, M.; Hannan, N.; Millan, E.; Smithers, L.E.; Trotter, M.; Rugg-Gunn, P.; Weber, A.; et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS ONE 2009, 4, e6082. [Google Scholar] [CrossRef]
- Vallier, L.; Touboul, T.; Brown, S.; Cho, C.; Bilican, B.; Alexander, M.; Cedervall, J.; Chandran, S.; Ahrlund-Richter, L.; Weber, A.; et al. Signaling pathways controlling pluripotency and early cell fate decisions of human induced pluripotent stem cells. Stem Cells 2009, 27, 2655–2666. [Google Scholar] [CrossRef]
- Fukuta, M.; Nakai, Y.; Kirino, K.; Nakagawa, M.; Sekiguchi, K.; Nagata, S.; Matsumoto, Y.; Yamamoto, T.; Umeda, K.; Heike, T.; et al. Derivation of mesenchymal stromal cells from pluripotent stem cells through a neural crest lineage using small molecule compounds with defined media. PLoS ONE 2014, 9, e112291. [Google Scholar] [CrossRef]
- Halaidych, O.V.; Cochrane, A.; van den Hil, F.E.; Mummery, C.L.; Orlova, V.V. Quantitative Analysis of Intracellular Ca2+ Release and Contraction in hiPSC-Derived Vascular Smooth Muscle Cells. Stem Cell Rep. 2019, 12, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.L.; Kimelman, D. Regulation of canonical Wnt signaling by Brachyury is essential for posterior mesoderm formation. Dev. Cell 2008, 15, 121–133. [Google Scholar] [CrossRef]
- Takada, S.; Stark, K.L.; Shea, M.J.; Vassileva, G.; McMahon, J.A.; McMahon, A.P. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994, 8, 174–189. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Fujimori, T.; McMahon, A.P.; Takada, S. Evidence that absence of Wnt-3a signaling promotes neuralization instead of paraxial mesoderm development in the mouse. Dev. Biol. 1997, 183, 234–242. [Google Scholar] [CrossRef]
- Nostro, M.C.; Cheng, X.; Keller, G.M.; Gadue, P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell 2008, 2, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Green, J.B.; New, H.V.; Smith, J.C. Responses of embryonic Xenopus cells to activin and FGF are separated by multiple dose thresholds and correspond to distinct axes of the mesoderm. Cell 1992, 71, 731–739. [Google Scholar] [CrossRef]
- Mendjan, S.; Mascetti, V.L.; Ortmann, D.; Ortiz, M.; Karjosukarso, D.W.; Ng, Y.; Moreau, T.; Pedersen, R.A. NANOG and CDX2 pattern distinct subtypes of human mesoderm during exit from pluripotency. Cell Stem Cell 2014, 15, 310–325. [Google Scholar] [CrossRef]
- Beddington, R.S.; Rashbass, P.; Wilson, V. Brachyury—A gene affecting mouse gastrulation and early organogenesis. Dev. Suppl. 1992, 157–165. [Google Scholar] [CrossRef]
- Rivera-Perez, J.A.; Magnuson, T. Primitive streak formation in mice is preceded by localized activation of Brachyury and Wnt3. Dev. Biol. 2005, 288, 363–371. [Google Scholar] [CrossRef]
- Wilkinson, D.G.; Bhatt, S.; Herrmann, B.G. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature 1990, 343, 657–659. [Google Scholar] [CrossRef]
- Iyer, D.; Gambardella, L.; Bernard, W.G.; Serrano, F.; Mascetti, V.L.; Pedersen, R.A.; Talasila, A.; Sinha, S. Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 2015, 142, 1528–1541. [Google Scholar] [CrossRef]
- Granata, A.; Serrano, F.; Bernard, W.G.; McNamara, M.; Low, L.; Sastry, P.; Sinha, S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97–109. [Google Scholar] [CrossRef]
- Yang, L.; Geng, Z.; Nickel, T.; Johnson, C.; Gao, L.; Dutton, J.; Hou, C.; Zhang, J. Differentiation of Human Induced-Pluripotent Stem Cells into Smooth-Muscle Cells: Two Novel Protocols. PLoS ONE 2016, 11, e0147155. [Google Scholar] [CrossRef]
- Kwong, G.; Marquez, H.A.; Yang, C.; Wong, J.Y.; Kotton, D.N. Generation of a Purified iPSC-Derived Smooth Muscle-like Population for Cell Sheet Engineering. Stem Cell Rep. 2019, 13, 499–514. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Marchand, M.; Anderson, E.K.; Phadnis, S.M.; Longaker, M.T.; Cooke, J.P.; Chen, B.; Reijo Pera, R.A. Concurrent generation of functional smooth muscle and endothelial cells via a vascular progenitor. Stem Cells Transl. Med. 2014, 3, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Lian, X.; Dunn, K.K.; Shi, M.; Han, T.; Qian, T.; Bhute, V.J.; Canfield, S.G.; Palecek, S.P. Chemically-defined albumin-free differentiation of human pluripotent stem cells to endothelial progenitor cells. Stem Cell Res. 2015, 15, 122–129. [Google Scholar] [CrossRef]
- Bajpai, V.K.; Mistriotis, P.; Loh, Y.H.; Daley, G.Q.; Andreadis, S.T. Functional vascular smooth muscle cells derived from human induced pluripotent stem cells via mesenchymal stem cell intermediates. Cardiovasc. Res. 2012, 96, 391–400. [Google Scholar] [CrossRef][Green Version]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.W.; Thomson, J.A.; Slukvin, I.I. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef]
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef]
- Uenishi, G.; Theisen, D.; Lee, J.H.; Kumar, A.; Raymond, M.; Vodyanik, M.; Swanson, S.; Stewart, R.; Thomson, J.; Slukvin, I. Tenascin C promotes hematoendothelial development and T lymphoid commitment from human pluripotent stem cells in chemically defined conditions. Stem Cell Rep. 2014, 3, 1073–1084. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Rasmussen, L.M.; Wolf, Y.G.; Ruoslahti, E. Vascular smooth muscle cells from injured rat aortas display elevated matrix production associated with transforming growth factor-beta activity. Am. J. Pathol. 1995, 147, 1041–1048. [Google Scholar]
- Karamariti, E.; Margariti, A.; Winkler, B.; Wang, X.; Hong, X.; Baban, D.; Ragoussis, J.; Huang, Y.; Han, J.D.; Wong, M.M.; et al. Smooth muscle cells differentiated from reprogrammed embryonic lung fibroblasts through DKK3 signaling are potent for tissue engineering of vascular grafts. Circ. Res. 2013, 112, 1433–1443. [Google Scholar] [CrossRef]
- Xiao, Q.; Zeng, L.; Zhang, Z.; Hu, Y.; Xu, Q. Stem cell-derived Sca-1+ progenitors differentiate into smooth muscle cells, which is mediated by collagen IV-integrin alpha1/beta1/alphav and PDGF receptor pathways. Am. J. Physiol. Cell Physiol. 2007, 292, C342–C352. [Google Scholar] [CrossRef]
- Lindahl, P.; Johansson, B.R.; Leveen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [CrossRef]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [CrossRef]
- Owens, G.K.; Geisterfer, A.A.; Yang, Y.W.; Komoriya, A. Transforming growth factor-beta-induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J. Cell Biol. 1988, 107, 771–780. [Google Scholar] [CrossRef]
- Bjorkerud, S. Effects of transforming growth factor-beta 1 on human arterial smooth muscle cells in vitro. Arterioscler. Thromb. 1991, 11, 892–902. [Google Scholar] [CrossRef]
- Deaton, R.A.; Su, C.; Valencia, T.G.; Grant, S.R. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J. Biol. Chem. 2005, 280, 31172–31181. [Google Scholar] [CrossRef]
- Servant, M.J.; Giasson, E.; Meloche, S. Inhibition of growth factor-induced protein synthesis by a selective MEK inhibitor in aortic smooth muscle cells. J. Biol. Chem. 1996, 271, 16047–16052. [Google Scholar] [CrossRef]
- Dash, B.C.; Levi, K.; Schwan, J.; Luo, J.; Bartulos, O.; Wu, H.; Qiu, C.; Yi, T.; Ren, Y.; Campbell, S.; et al. Tissue-Engineered Vascular Rings from Human iPSC-Derived Smooth Muscle Cells. Stem Cell Rep. 2016, 7, 19–28. [Google Scholar] [CrossRef]
- Wanjare, M.; Kuo, F.; Gerecht, S. Derivation and maturation of synthetic and contractile vascular smooth muscle cells from human pluripotent stem cells. Cardiovasc. Res. 2013, 97, 321–330. [Google Scholar] [CrossRef]
- Wanjare, M.; Kusuma, S.; Gerecht, S. Defining differences among perivascular cells derived from human pluripotent stem cells. Stem Cell Rep. 2014, 2, 561–575. [Google Scholar] [CrossRef]
- Frid, M.G.; Shekhonin, B.V.; Koteliansky, V.E.; Glukhova, M.A. Phenotypic changes of human smooth muscle cells during development: Late expression of heavy caldesmon and calponin. Dev. Biol. 1992, 153, 185–193. [Google Scholar] [CrossRef]
- Frid, M.G.; Moiseeva, E.P.; Stenmark, K.R. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ. Res. 1994, 75, 669–681. [Google Scholar] [CrossRef]
- Glukhova, M.A.; Frid, M.G.; Koteliansky, V.E. Phenotypic changes of human aortic smooth muscle cells during development and in adult. J. Atheroscler. Thromb. 1994, 1 (Suppl. 1), S47–S49. [Google Scholar] [CrossRef] [PubMed]
- Villaschi, S.; Nicosia, R.F.; Smith, M.R. Isolation of a morphologically and functionally distinct smooth muscle cell type from the intimal aspect of the normal rat aorta. Evidence for smooth muscle cell heterogeneity. Vitr. Cell. Dev. Biol. Anim. 1994, 30A, 589–595. [Google Scholar] [CrossRef]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Arsenian, S.; Weinhold, B.; Oelgeschlager, M.; Ruther, U.; Nordheim, A. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 1998, 17, 6289–6299. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.Z.; Wang, Z.; Richardson, J.A.; Olson, E.N. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 2003, 100, 9366–9370. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.Z.; Pipes, G.C.; Olson, E.N. Myocardin is a master regulator of smooth muscle gene expression. Proc. Natl. Acad. Sci. USA 2003, 100, 7129–7134. [Google Scholar] [CrossRef]
- Yoshida, T.; Sinha, S.; Dandre, F.; Wamhoff, B.R.; Hoofnagle, M.H.; Kremer, B.E.; Wang, D.Z.; Olson, E.N.; Owens, G.K. Myocardin is a key regulator of CArG-dependent transcription of multiple smooth muscle marker genes. Circ. Res. 2003, 92, 856–864. [Google Scholar] [CrossRef]
- Wang, D.Z.; Li, S.; Hockemeyer, D.; Sutherland, L.; Wang, Z.; Schratt, G.; Richardson, J.A.; Nordheim, A.; Olson, E.N. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14855–14860. [Google Scholar] [CrossRef]
- Chang, D.F.; Belaguli, N.S.; Iyer, D.; Roberts, W.B.; Wu, S.P.; Dong, X.R.; Marx, J.G.; Moore, M.S.; Beckerle, M.C.; Majesky, M.W.; et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev. Cell 2003, 4, 107–118. [Google Scholar] [CrossRef]
- Shang, Y.; Yoshida, T.; Amendt, B.A.; Martin, J.F.; Owens, G.K. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. J. Cell Biol. 2008, 181, 461–473. [Google Scholar] [CrossRef]
- Clayton, Z.E.; Yuen, G.S.; Sadeghipour, S.; Hywood, J.D.; Wong, J.W.; Huang, N.F.; Ng, M.K.; Cooke, J.P.; Patel, S. A comparison of the pro-angiogenic potential of human induced pluripotent stem cell derived endothelial cells and induced endothelial cells in a murine model of peripheral arterial disease. Int. J. Cardiol. 2017, 234, 81–89. [Google Scholar] [CrossRef]
- Lai, W.H.; Ho, J.C.; Chan, Y.C.; Ng, J.H.; Au, K.W.; Wong, L.Y.; Siu, C.W.; Tse, H.F. Attenuation of hind-limb ischemia in mice with endothelial-like cells derived from different sources of human stem cells. PLoS ONE 2013, 8, e57876. [Google Scholar] [CrossRef]
- Mulyasasmita, W.; Cai, L.; Dewi, R.E.; Jha, A.; Ullmann, S.D.; Luong, R.H.; Huang, N.F.; Heilshorn, S.C. Avidity-controlled hydrogels for injectable co-delivery of induced pluripotent stem cell-derived endothelial cells and growth factors. J. Control. Release 2014, 191, 71–81. [Google Scholar] [CrossRef]
- Foster, A.A.; Dewi, R.E.; Cai, L.; Hou, L.; Strassberg, Z.; Alcazar, C.A.; Heilshorn, S.C.; Huang, N.F. Protein-engineered hydrogels enhance the survival of induced pluripotent stem cell-derived endothelial cells for treatment of peripheral arterial disease. Biomater. Sci. 2018, 6, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Nguyen, P.K.; Lee, A.S.; Xu, D.; Hu, S.; Plews, J.R.; Han, L.; Huber, B.C.; Lee, W.H.; Gong, Y.; et al. Microfluidic single-cell analysis shows that porcine induced pluripotent stem cell-derived endothelial cells improve myocardial function by paracrine activation. Circ. Res. 2012, 111, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.; Domev, H.; Ben-Yosef, O.; Tzukerman, M.; Zeevi-Levin, N.; Novak, A.; Germanguz, I.; Amit, M.; Itskovitz-Eldor, J. Multipotent vasculogenic pericytes from human pluripotent stem cells promote recovery of murine ischemic limb. Circulation 2012, 125, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Gao, M.; Gorecka, J.; Langford, J.; Liu, J.; Luo, J.; Taniguchi, R.; Matsubara, Y.; Liu, H.; Guo, L.; et al. Human-Induced Pluripotent Stem-Cell-Derived Smooth Muscle Cells Increase Angiogenesis to Treat Hindlimb Ischemia. Cells 2021, 10, 792. [Google Scholar] [CrossRef]
- Atchison, L.; Zhang, H.; Cao, K.; Truskey, G.A. A Tissue Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome Using Human iPSC-derived Smooth Muscle Cells. Sci. Rep. 2017, 7, 8168. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Qin, L.; Zhao, L.; Gui, L.; Ellis, M.W.; Huang, Y.; Kural, M.H.; Clark, J.A.; Ono, S.; Wang, J.; et al. Tissue-Engineered Vascular Grafts with Advanced Mechanical Strength from Human iPSCs. Cell Stem Cell 2020, 26, 251–261 e258. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Y.; Jiao, J.; Liu, Z.; Zhao, C.; Zhou, Z.; Zhang, Z.; Forde, K.; Wang, L.; Wang, J.; et al. Patient-specific cardiovascular progenitor cells derived from integration-free induced pluripotent stem cells for vascular tissue regeneration. Biomaterials 2015, 73, 51–59. [Google Scholar] [CrossRef]
- Luo, J.; Lin, Y.; Shi, X.; Li, G.; Kural, M.H.; Anderson, C.W.; Ellis, M.W.; Riaz, M.; Tellides, G.; Niklason, L.E.; et al. Xenogeneic-free generation of vascular smooth muscle cells from human induced pluripotent stem cells for vascular tissue engineering. Acta Biomater. 2021, 119, 155–168. [Google Scholar] [CrossRef]
- Li, Z.; Hu, S.; Ghosh, Z.; Han, Z.; Wu, J.C. Functional characterization and expression profiling of human induced pluripotent stem cell- and embryonic stem cell-derived endothelial cells. Stem Cells Dev. 2011, 20, 1701–1710. [Google Scholar] [CrossRef]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.; Dzeja, P.P.; Nelson, T.J.; Terzic, A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012, 11, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Deinsberger, J.; Reisinger, D.; Weber, B. Global trends in clinical trials involving pluripotent stem cells: A systematic multi-database analysis. NPJ Regen. Med. 2020, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Heslop, J.A.; Hammond, T.G.; Santeramo, I.; Tort Piella, A.; Hopp, I.; Zhou, J.; Baty, R.; Graziano, E.I.; Proto Marco, B.; Caron, A.; et al. Concise review: Workshop review: Understanding and assessing the risks of stem cell-based therapies. Stem Cells Transl. Med. 2015, 4, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.; Tsuzuki, S.; Yasui, R.; Kobayashi, T.; Ikeda, K.; Hamada, Y.; Kanai, E.; Camp, J.G.; Treutlein, B.; Ueno, Y.; et al. Robust detection of undifferentiated iPSC among differentiated cells. Sci. Rep. 2020, 10, 10293. [Google Scholar] [CrossRef]
- Gutierrez-Aranda, I.; Ramos-Mejia, V.; Bueno, C.; Munoz-Lopez, M.; Real, P.J.; Macia, A.; Sanchez, L.; Ligero, G.; Garcia-Parez, J.L.; Menendez, P. Human induced pluripotent stem cells develop teratoma more efficiently and faster than human embryonic stem cells regardless the site of injection. Stem Cells 2010, 28, 1568–1570. [Google Scholar] [CrossRef]
- Kot, M.; Baj-Krzyworzeka, M.; Szatanek, R.; Musial-Wysocka, A.; Suda-Szczurek, M.; Majka, M. The Importance of HLA Assessment in “Off-the-Shelf” Allogeneic Mesenchymal Stem Cells Based-Therapies. Int. J. Mol. Sci. 2019, 20, 5680. [Google Scholar] [CrossRef]
- Creane, M.; Howard, L.; O’Brien, T.; Coleman, C.M. Biodistribution and retention of locally administered human mesenchymal stromal cells: Quantitative polymerase chain reaction-based detection of human DNA in murine organs. Cytotherapy 2017, 19, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Zhao, C.; Wu, S.; Zhang, C.; Liu, H.; Fan, Y. Hydrogel-based therapeutic angiogenesis: An alternative treatment strategy for critical limb ischemia. Biomaterials 2021, 274, 120872. [Google Scholar] [CrossRef]
- Li, R.K.; Jia, Z.Q.; Weisel, R.D.; Merante, F.; Mickle, D.A. Smooth muscle cell transplantation into myocardial scar tissue improves heart function. J. Mol. Cell. Cardiol. 1999, 31, 513–522. [Google Scholar] [CrossRef]
- Sakai, T.; Li, R.K.; Weisel, R.D.; Mickle, D.A.; Jia, Z.Q.; Tomita, S.; Kim, E.J.; Yau, T.M. Fetal cell transplantation: A comparison of three cell types. J. Thorac. Cardiovasc. Surg. 1999, 118, 715–724. [Google Scholar] [CrossRef][Green Version]
- Fujii, T.; Yau, T.M.; Weisel, R.D.; Ohno, N.; Mickle, D.A.; Shiono, N.; Ozawa, T.; Matsubayashi, K.; Li, R.K. Cell transplantation to prevent heart failure: A comparison of cell types. Ann. Thorac. Surg. 2003, 76, 2062–2070; discussion 2070. [Google Scholar] [CrossRef]
- Liu, T.B.; Fedak, P.W.; Weisel, R.D.; Yasuda, T.; Kiani, G.; Mickle, D.A.; Jia, Z.Q.; Li, R.K. Enhanced IGF-1 expression improves smooth muscle cell engraftment after cell transplantation. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2840–H2849. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yasuda, T.; Weisel, R.D.; Li, R.K. Enhanced cell transplantation: Preventing apoptosis increases cell survival and ventricular function. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H939–H947. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.J.; Li, R.K.; Weisel, R.D.; Mickle, D.A.; Li, G.; Yau, T.M. Autologous smooth muscle cell transplantation improved heart function in dilated cardiomyopathy. Ann. Thorac. Surg. 2000, 70, 859–865. [Google Scholar] [CrossRef]
- Yoo, K.J.; Li, R.K.; Weisel, R.D.; Mickle, D.A.; Tomita, S.; Ohno, N.; Fujii, T. Smooth muscle cells transplantation is better than heart cells transplantation for improvement of heart function in dilated cardiomyopathy. Yonsei Med. J. 2002, 43, 296–303. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Methods | Expected Features |
|---|---|---|
| Cell morphology | Bright-field/phase-contrast microscopy | Cobblestone-like shape with a single nucleus |
| Increased expression of EC-specific markers | RT-PCR Western blot Flow cytometry Immunocytochemistry | KDR, CDH5, VWF, PECAM1, TEK, and NOS3 |
| In silico analysis | Microarray RNA-sequencing Single-cell RNA-sequencing Single-cell ATAC-sequencing | Upregulation of EC-specific genes Enriched GO terms related to ECs Comparison with human primary ECs |
| In vitro functionality | Immunocytochemistry | NO production Uptake of acetylated LDL |
| RT-PCR Western blot ELISA | Elevated expression of angiogenic factors, including VEGFA, ANGPT1, IGF1, and FGF2 | |
| Cell migration | Increased overall motility | |
| Tube formation | Tube-like structure formation |
| Characteristics | Methods | Expected Features |
|---|---|---|
| Cell morphology | Bright-field/phase-contrast microscopy Transmission electron microscopy | Spindle-shaped with a single nucleus Enhanced filamentous patterns and dense bodies |
| Increased expression of contractile SMC-specific markers | RT-PCR Western blot Flow cytometry Immunocytochemistry | ACTA2, TAGLN, CALD1, DES, CNN1, MYH11, and SMTN |
| Reporter transgene | Contractile SMC-specific gene promoter | |
| (Optional) Detection of contractile SMC fate determining transcription factors | RT-PCR Western blot Flow cytometry Immunocytochemistry | SRF, MYOCD, MRTFA and B, CRIP1 and 2, and PITX2 |
| Transcriptome analysis | Microarray RNA-sequencing Single-cell RNA-sequencing | Upregulation of contractile SMC-specific genes Enriched GO terms related to contractile SMCs Comparison with primary human SMCs |
| In vitro functionality | Contraction Intracellular calcium release Force generation | Responding to vasoactive agents |
| Cell Type | Number of Cells/Head | Delivery Method | Species (Sex) | Animal Model | Reference |
|---|---|---|---|---|---|
| hiPSC-derived EC | 5 × 105 | Intramuscular injection | NOD/SCID mouse (male) | Hindlimb ischemia | [70] |
| 1 × 106 | Intramuscular injection | NOD/SCID mouse (male) | Hindlimb ischemia | [234] | |
| 3 × 106 | Intramuscular injection | SCID mouse (male) | Hindlimb ischemia | [235] | |
| 5 × 105 | Intramuscular injection | NOD/SCID mouse (male) | Hindlimb ischemia | [236] | |
| 1 × 106 | Intramuscular injection | NOD/SCID mouse (male) | Hindlimb ischemia | [237] | |
| 2 × 105 | Intramuscular injection | Athymic nude Foxn1nu mouse (male) | Hindlimb ischemia | [73] | |
| 1 × 106 | Intramyocardial injection | NOD/SCID mouse (female) | Myocardial infarction | [238] * | |
| hiPSC-derived pericyte | 2 × 106 | Intramuscular injection | CD-1 nude mouse (male) | Hindlimb ischemia | [239] |
| hiPSC-derived SMC | 1 × 106 | Intramuscular injection | Athymic nude Foxn1nu mouse (male) | Hindlimb ischemia | [240] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.E.; Jung, C.; Yoon, Y.-s. Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions. J. Cardiovasc. Dev. Dis. 2021, 8, 148. https://doi.org/10.3390/jcdd8110148
Oh JE, Jung C, Yoon Y-s. Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions. Journal of Cardiovascular Development and Disease. 2021; 8(11):148. https://doi.org/10.3390/jcdd8110148
Chicago/Turabian StyleOh, Jee Eun, Cholomi Jung, and Young-sup Yoon. 2021. "Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions" Journal of Cardiovascular Development and Disease 8, no. 11: 148. https://doi.org/10.3390/jcdd8110148
APA StyleOh, J. E., Jung, C., & Yoon, Y.-s. (2021). Human Induced Pluripotent Stem Cell-Derived Vascular Cells: Recent Progress and Future Directions. Journal of Cardiovascular Development and Disease, 8(11), 148. https://doi.org/10.3390/jcdd8110148