Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts


Abstract
1. Introduction
2. Cardiac Fibroblasts and the Extracellular Matrix
3. Contributions of Cardiac Fibroblasts and the ECM to Cardiac Regeneration in Urodeles
4. Contributions of Cardiac Fibroblasts and the ECM to Cardiac Regeneration in Teleosts
5. Contributions of Cardiac Fibroblasts and the ECM to Cardiac Regeneration in Mammals
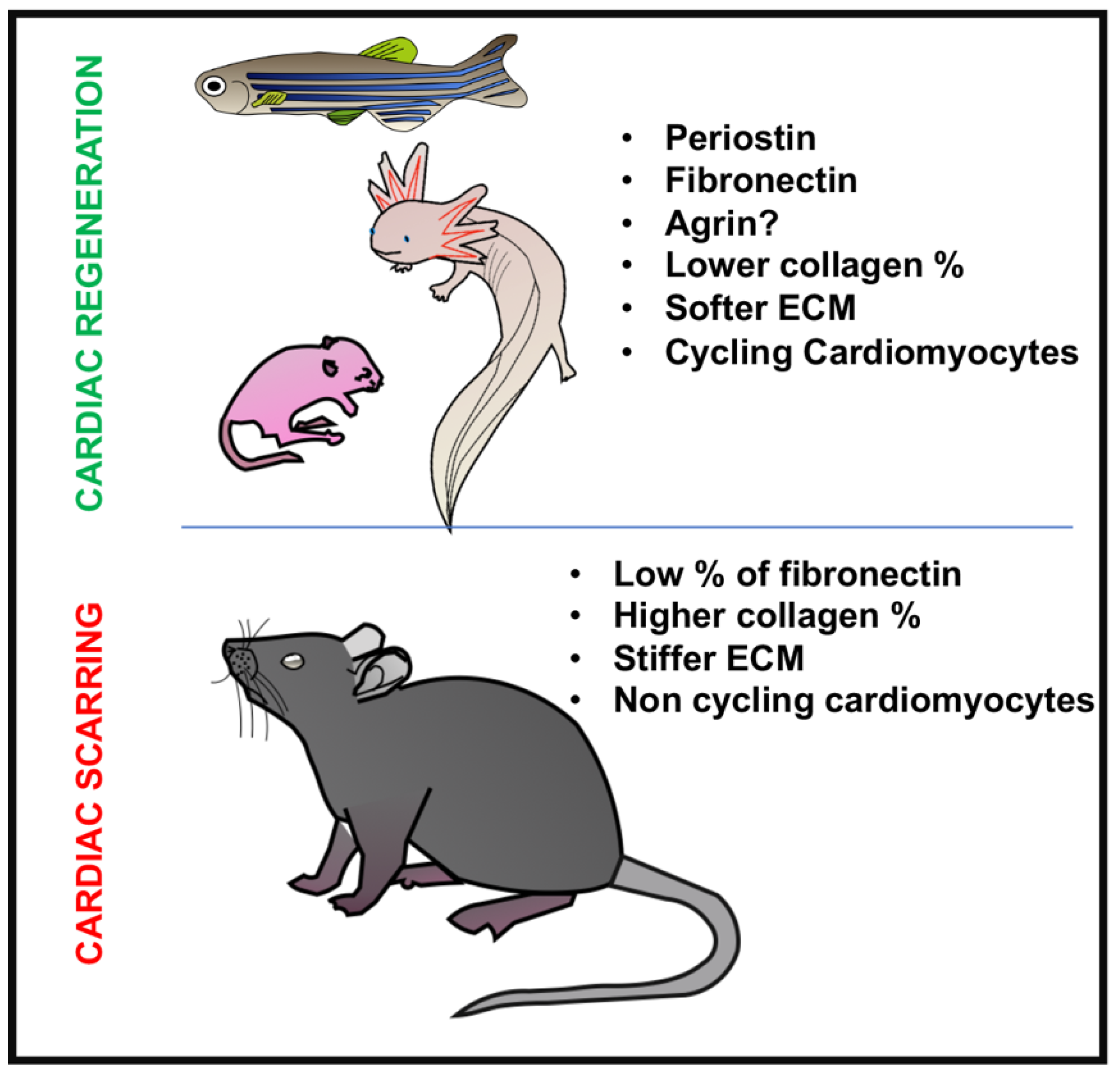
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| TGF-β | Transforming growth factor-β |
| BMP | Bone morphogenic protein |
| Wnt | Wingless-related integration site |
| b-HLH | Basic helix-loop-helix |
| Tcf21 | Transcription factor 21 |
| PDGFR-α | Platelet-derived growth factor receptor alpha |
| FN | Fibronectin |
| Postn | Periostin |
| MMPs | Matrix metalloproteinases |
| HA | Hyaluronic acid |
| TNC | Tenascin-C |
| Fg | Fibrinogen |
| Col | Collagen |
References
- Tanaka, E.M.; Reddien, P.W. The cellular basis for animal regeneration. Dev. Cell 2011, 21, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Uygur, A.; Lee, R.T. Mechanisms of Cardiac Regeneration. Dev. Cell 2016, 36, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.; Sleep, E.; Raya, M.; Martí, M.; Raya, A.; Belmonte, J.C.I. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; López, B.; Coelho-Filho, O.R.; Lakdawala, N.K.; Cirino, A.L.; Jarolim, P.; Kwong, R.; Gonzalez, A.; Colan, S.D.; Seidman, J.; et al. Myocardial Fibrosis as an Early Manifestation of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2010, 363, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.H.; Ismail, N.A.; Dweck, M.R.; et al. Association of Fibrosis With Mortality and Sudden Cardiac Death in Patients with Nonischemic Dilated Cardiomyopathy. JAMA 2013, 309, 896. [Google Scholar] [CrossRef] [PubMed]
- Schelbert, E.B.; Fridman, Y.; Wong, T.C.; Abu Daya, H.; Piehler, K.M.; Kadakkal, A.; Miller, C.A.; Ugander, M.; Maanja, M.; Kellman, P.; et al. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: Association with baseline disease severity and subsequent outcome. JAMA Cardiol. 2017, 2, 995–1006. [Google Scholar] [CrossRef]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial derived cell epithelial to mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Simões, F.C.; Riley, P.R. The ontogeny, activation and function of the epicardium during heart development and regeneration. Development 2018, 145, dev155994. [Google Scholar] [CrossRef]
- Wessels, A.; Hoff, M.J.B.V.D.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; Van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially-derived Fibroblasts Preferentially Contribute to the Parietal Leaflets of the Atrioventricular Valves in the Murine Heart. Dev. Boil. 2012, 366, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Dettman, R.W.; Denetclaw, W.; Ordahl, C.P.; Bristow, J. Common Epicardial Origin of Coronary Vascular Smooth Muscle, Perivascular Fibroblasts, and Intermyocardial Fibroblasts in the Avian Heart. Dev. Boil. 1998, 193, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, T.; Gourdie, R.G. Pericardial Mesoderm Generates a Population of Coronary Smooth Muscle Cells Migrating into the Heart along with Ingrowth of the Epicardial Organ. Dev. Boil. 1996, 174, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, T.; Fischman, D.A. Retroviral analysis of cardiac morphogenesis: Discontinuous formation of coronary vessels. Proc. Natl. Acad. Sci. USA 1992, 89, 9504–9508. [Google Scholar] [CrossRef] [PubMed]
- Groot, A.C.G.-D.; Peeters, M.-P.F.V.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E.; Peeters, M.-P.F.V. Epicardium-Derived Cells Contribute a Novel Population to the Myocardial Wall and the Atrioventricular Cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial- and Endothelial- to Mesenchymal Transition: From Cardiovascular Development to Disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef]
- Braitsch, C.M.; Yutzey, K.E. Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells. J. Dev. Boil. 2013, 1, 92–111. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell. Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef]
- Ivey, M.J.; Kuwabara, J.T.; Pai, J.T.; Moore, R.E.; Sun, Z.; Tallquist, M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell. Cardiol. 2018, 114, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [PubMed]
- Velnar, T.; Bailey, T.; Smrkolj, V. The Wound Healing Process: An Overview of the Cellular and Molecular Mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair and remodeling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Stretching the boundaries of extracellular matrix research. Nat. Rev. Mol. Cell Boil. 2014, 15, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Mundell, N.A.; Jessen, J.R. Extracellular Matrix Remodeling in Zebrafish Development. In Extracellular Matrix in Development; De Simone, D., Mecham, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Jessen, J.R. Recent advances in the study of zebrafish extracellular matrix proteins. Dev. Boil. 2015, 401, 110–121. [Google Scholar] [CrossRef]
- Byron, A.; Humphries, J.D.; Humphries, M.J. Defining the extracellular matrix using proteomics. Int. J. Exp. Pathol. 2013, 94, 75–92. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Iyer, R.P.; Jung, M.; DeLeon-Pennell, K.Y.; Ma, Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J. Mol. Cell. Cardiol. 2016, 91, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta BBA 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.O.; Chapin, S.; Sherry, R. Regeneration of the ventricular myocardium in amphibians. Nature 1974, 248, 145–147. [Google Scholar] [CrossRef]
- McDonnell, T.J.; Oberpriller, J.O. The atrial proliferative response following partial ventricular amputation in the heart of the adult newt. A light and electron microscopic autoradiographic study. Tissue Cell 1983, 15, 351–363. [Google Scholar] [CrossRef]
- McDonnell, T.J.; Oberpriller, J.O. The response of the atrium to direct mechanical wounding in the adult heart of the newt, Notophthalmus viridescens. An electron-microscopic and autoradiographic study. Cell Tissue Res. 1984, 235, 583–592. [Google Scholar] [CrossRef]
- Oberpriller, J.O.; Oberpriller, J.C.; Matz, D.G.; Soonpaa, M.H. Stimulation of Proliferative Events in the Adult Amphibian Cardiac Myocyte. Ann. N. Y. Acad. Sci. 1995, 752, 30–46. [Google Scholar] [CrossRef]
- Flink, I.L. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: Confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Brain Struct. Funct. 2002, 205, 235–244. [Google Scholar]
- Cano-Martínez, A.; Vargas-González, A.; Guarner-Lans, V.; Prado-Zayago, E.; León-Oleda, M.; Nieto-Lima, B. Functional and structural regeneration in the axolotl heart (Ambystoma mexicanum) after partial ventricular amputation. Arch. Cardiol. Mexico 2010, 80, 79–86. [Google Scholar]
- Oberpriller, J.O.; Oberpriller, J.C. Response of the adult newt ventricle to injury. J. Exp. Zool. 1974, 187, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Witman, N.; Murtuza, B.; Davis, B.; Arner, A.; Morrison, J.I. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev. Boil. 2011, 354, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.E.; Odelberg, S.J.; Simon, H.-G. A Dynamic Spatiotemporal Extracellular Matrix Facilitates Epicardial-Mediated Vertebrate Heart Regeneration. Dev. Boil. 2013, 382, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Piatkowski, T.; Mühlfeld, C.; Borchardt, T.; Braun, T. Reconstitution of the Myocardium in Regenerating Newt Hearts is Preceded by Transient Deposition of Extracellular Matrix Components. Stem Cells Dev. 2013, 22, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Magadum, A.; Engel, F.B. Cardiomyocyte proliferation in cardiac development and regeneration: A guide to methodologies and interpretations. Am. J. Physiol. Circ. Physiol. 2015, 309, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Godwin, J.W.; Debuque, R.; Salimova, E.; Rosenthal, N.A. Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen. Med. 2017, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.K.; Zalewski, A.A.; Reddi, A. An immunofluorescent study of the distribution of fibronectin and laminin during limb regeneration in the adult newt. Dev. Boil. 1983, 96, 355–365. [Google Scholar] [CrossRef]
- Mescher, A.L.; Munaim, S.I. Changes in the extracellular matrix and glycosaminoglycan synthesis during the initiation of regeneration in adult newt forelimbs. Anat. Rec. Adv. Integr. Anat. Evol. Boil. 1986, 214, 424–431. [Google Scholar] [CrossRef]
- Onda, H.; Poulin, M.L.; Tassava, R.A.; Chiu, I.-M. Characterization of a newt tenascin cDNA and localization of tenascin mRNA during newt limb regeneration by in situ hybridization. Dev. Boil. 1991, 148, 219–232. [Google Scholar] [CrossRef]
- Asahina, K.; Obara, M.; Yoshizato, K. Expression of genes of type I and type II collagen in the formation and development of the blastema of regenerating newt limb. Dev. Dyn. 1999, 216, 59–71. [Google Scholar] [CrossRef]
- Vinarsky, V.; Atkinson, D.L.; Stevenson, T.J.; Keating, M.T.; Odelberg, S.J. Normal newt limb regeneration requires matrix metalloproteinase function. Dev. Boil. 2005, 279, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Bakkers, J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc. Res. 2011, 91, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A Dynamic Epicardial Injury Response Supports Progenitor Cell Activity during Zebrafish Heart Regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef]
- Sánchez-Iranzo, H.; Galardi-Castilla, M.; Sanz-Morejón, A.; González-Rosa, J.M.; Costa, R.; Ernst, A.; De Aja, J.S.; Langa, X.; Mercader, N. Transient fibrosis resolves via fibroblast inactivation in the regenerating zebrafish heart. Proc. Natl. Acad. Sci. USA 2018, 115, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Navis, A.; Cox, B.D.; Dickson, A.L.; Gemberling, M.; Karra, R.; Bagnat, M.; Poss, K.D. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 2016, 143, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Xiang, F.-L.; Fang, M.; Yutzey, K.E. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. [Google Scholar] [CrossRef]
- Stockdale, W.T.; Lemieux, M.E.; Killen, A.C.; Zhao, J.; Hu, Z.; Riepsaame, J.; Hamilton, N.; Kudoh, T.; Riley, P.R.; Van Aerle, R.; et al. Heart Regeneration in the Mexican Cavefish. Cell Rep. 2018, 25, 1997–2007. [Google Scholar] [CrossRef]
- Ito, K.; Morioka, M.; Kimura, S.; Tasaki, M.; Inohaya, K.; Kudo, A. Differential reparative phenotypes between zebrafish and medaka after cardiac injury. Dev. Dyn. 2014, 243, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Trinh, L.A.; Stainier, D.Y. Cardiac development. Methods Cell Biol. 2004, 76, 455–473. [Google Scholar] [PubMed]
- Sakaguchi, T.; Kikuchi, Y.; Kuroiwa, A.; Takeda, H.; Stainier, D.Y.R. The yolk syncytial layer regulates myocardial migration by influencing extracellular matrix assembly in zebrafish. Development 2006, 133, 4063–4072. [Google Scholar] [CrossRef] [PubMed]
- Arrington, C.B.; Yost, H.J. Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development 2009, 136, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Langenbacher, A.D.; Huang, J.; Chen, Y.; Chen, J.N. Sodium pump activity in the yolk syncytial layer regulates zebrafish heart tube morphogenesis. Dev. Biol. 2012, 362, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Konstandin, M.H.; Toko, H.; Gastelum, G.M.; Quijada, P.; De La Torre, A.; Quintana, M.; Collins, B.; Din, S.; Avitabile, D.; Völkers, M.; et al. Fibronectin is Essential for Reparative Cardiac Progenitor Cell Response Following Myocardial Infarction. Circ. Res. 2013, 113, 115–125. [Google Scholar] [CrossRef]
- Garcia-Puig, A.; Mosquera, J.L.; Jimenez-Delgado, S.; Garcia-Pastor, C.; Jorba, I.; Navajas, D.; Canals, G.; Raya, A. Proteomics analysis of extracellular matrix remodeling during zebrafish heart regeneration. Mol. Cell. Proteom. 2019. [Google Scholar] [CrossRef]
- Wang, J.; Karra, R.; Dickson, A.L.; Poss, K.D. Fibronectin is deposited by injury-activated epicardial cells and is necessary for zebrafish heart regeneration. Dev. Boil. 2013, 382, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGF signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; Mckean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A.; Banerjee, I. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Circ. Physiol. 2007, 293, 1883–1891. [Google Scholar]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; D’Agostino, G.; Loo, S.J.; Wang, C.X.; Su, L.P.; Tan, S.H.; Tee, G.Z.; Pua, C.J.; Pena, E.M.; Cheng, R.B.; et al. Early Regenerative Capacity in the Porcine Heart. Circulation 2018, 138, 2798–2808. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, E.; Zhao, M.; Chong, Z.; Fan, C.; Tang, Y.; Hunter, J.D.; Borovjagin, A.V.; Walcott, G.P.; Chen, J.Y.; et al. Regenerative Potential of Neonatal Porcine Hearts. Circulation 2018, 138, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Haubner, B.J.; Schneider, J.; Schweigmann, U.; Schuetz, T.; Dichtl, W.; Velik-Salchner, C.; Stein, J.I.; Penninger, J.M. Functional Recovery of a Human Neonatal Heart after Severe Myocardial Infarction. Circ. Res. 2016, 118, 216–221. [Google Scholar] [CrossRef]
- Moore, A.L.; Marshall, C.D.; Barnes, L.A.; Murphy, M.P.; Ransom, R.C.; Longaker, M.T. Scarless wound healing: Transitioning from fetal research to regenerative healing. Wiley Interdiscip. Rev. Dev. Boil. 2018, 7, e309. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. Intramyocardial Fibroblast—Myocyte Communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef]
- Jonsson, M.K.; Hartman, R.J.; Ackers-Johnson, M.; Tan, W.L.; Lim, B.; Van Veen, T.A.; Foo, R.S. A Transcriptomic and Epigenomic Comparison of Fetal and Adult Human Cardiac Fibroblasts Reveals Novel Key Transcription Factors in Adult Cardiac Fibroblasts. JACC Basic Transl. Sci. 2016, 1, 590–602. [Google Scholar] [CrossRef]
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.-T.; Shaw, R.M.; Srivastava, D. Cardiac Fibroblasts Regulate Myocardial Proliferation through β1 Integrin Signaling. Dev. Cell 2009, 16, 233–244. [Google Scholar] [CrossRef]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of Cardiac Fibroblasts and the Role of Periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Takefuji, M.; Ngai, C.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Möllmann, H.; et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ. Res. 2016, 118, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Ozhan, G.; Weidinger, G. Wnt/β-catenin signaling in heart regeneration. Cell Regen. 2015, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.J.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen producing cells in the injured heart. JCI Insight 2019, 4, e128722. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular Matrix and Heart Development. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Costell, M.; Carmona, R.; Gustafsson, E.; González-Iriarte, M.; Fässler, R.; Muñoz-Chápuli, R. Hyperplastic Conotruncal Endocardial Cushions and Transposition of Great Arteries in Perlecan-Null Mice. Circ. Res. 2002, 91, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, H.; Zhang, M.; Markwald, R.; Mjaatvedt, C. A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse. Dev. Boil. 1997, 186, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 2004, 230, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V Collagen Controls the Initiation of Collagen Fibril Assembly. J. Boil. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef] [PubMed]
- Georges-Labouesse, E.N.; George, E.L.; Rayburn, H.; Hynes, R.O. Mesodermal development in mouse embryos mutant for fibronectin. Dev. Dyn. 1996, 207, 145–156. [Google Scholar] [CrossRef]
- Wong, M.; Lawton, T.; Goetinck, P.F.; Kuhn, J.L.; Goldstein, S.A.; Bonadio, J. Aggrecan core protein is expressed in membranous bone of the chick embryo. Molecular and biomechanical studies of normal and nanomelia embryos. J. Boil. Chem. 1992, 267, 5592–5598. [Google Scholar]
- Watanabe, H.; Yamada, Y. Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nat. Genet. 1999, 21, 225–229. [Google Scholar] [CrossRef]
- Ng, A.; Wong, M.; Viviano, B.; Erlich, J.M.; Alba, G.; Pflederer, C.; Jay, P.Y.; Saunders, S. Loss of glypican-3 Function Causes Growth Factor-dependent Defects in Cardiac and Coronary Vascular Development. Dev. Boil. 2009, 335, 208–215. [Google Scholar] [CrossRef]
- Metsäranta, M. Chondrodysplasia in transgenic mice harboring a 15-amino acid deletion in the triple helical domain of pro alpha 1(II) collagen chain. J. Cell Boil. 1992, 118, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.B.; Twal, W.O.; Mjaatvedt, C.H.; Fairey, S.E.; Toole, B.P.; Iruela-Arispe, M.L.; Argraves, W.S. Proteolytic Cleavage of Versican During Cardiac Cushion Morphogenesis. Dev. Dyn. 2006, 235, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Dietz, H.C. Marfan’s syndrome. Lancet 2005, 366, 1965–1976. [Google Scholar] [CrossRef]
- Rios, H.; Koushik, S.V.; Wang, H.; Wang, J.; Zhou, H.-M.; Lindsley, A.; Rogers, R.; Chen, Z.; Maeda, M.; Kruzynska-Frejtag, A.; et al. periostin Null Mice Exhibit Dwarfism, Incisor Enamel Defects, and an Early-Onset Periodontal Disease-Like Phenotype. Mol. Cell. Boil. 2005, 25, 11131–11144. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Quinn, K.P.; Georgakoudi, I.; Black, L.D., 3rd. Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro. Acta Biomater. 2014, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Niebroj-Dobosz, I. Tenascin-C in human cardiac pathology. Clin. Chim. Acta 2012, 413, 1516–1518. [Google Scholar] [CrossRef]
- Kasprzycka, M.; Hammarström, C.; Haraldsen, G. Tenascins in fibrotic disorders—From bench to bedside. Cell Adhes. Migr. 2015, 9, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Kuhn, B.; Del Monte, F.; Hajjar, R.J.; Chang, Y.-S.; Lebeche, D.; Arab, S.; Keating, M.T. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 2007, 13, 962–969. [Google Scholar] [CrossRef]
- Lorts, A.; Schwanekamp, J.A.; Elrod, J.W.; Sargent, M.A.; Molkentin, J.D. Genetic manipulation of periostin expression in the heart does not affect myocyte content, cell cycle activity, or cardiac repair. Circ. Res. 2009, 104, e1–e7. [Google Scholar] [CrossRef]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Umansky, K.B.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Bassat, D.R.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yahalom-Ronen, Y.; Rajchman, D.; Sarig, R.; Geiger, B.; Tzahor, E. Reduced matrix rigidity promotes neonatal cardiomyocyte dedifferentiation, proliferation and clonal expansion. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Notari, M.; Ventura-Rubio, A.; Bedford-Guaus, S.J.; Jorba, I.; Mulero, L.; Navajas, D.; Martí, M.; Raya, A. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci. Adv. 2018, 4, eaao5553. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.L.; Ardehali, R. Direct Cardiac Reprogramming: Progress and Promise. Stem Cells Int. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Bjørnstad, J.L.; Skrbic, B.; Engebretsen, K.V.; Strand, M.E.; Lunde, I.G.; Herum, K.M.; Marstein, H.S.; Sjaastad, I.; Carlson, C.R.; Christensen, G.; et al. Lack of collagen VIII reduces fibrosis and promotes early mortality and cardiac dilatation in pressure overload in mice. Cardiovasc. Res. 2015, 106, 32–42. [Google Scholar]
- Marro, J.; Pfefferli, C.; Charles, A.-S.D.P.; Bise, T.; Jaźwińska, A. Collagen XII Contributes to Epicardial and Connective Tissues in the Zebrafish Heart during Ontogenesis and Regeneration. PLoS ONE 2016, 11, e0165497. [Google Scholar] [CrossRef]
- Snider, P.; Hinton, R.B.; Moreno-Rodriguez, R.A.; Wang, J.; Rogers, R.; Lindsley, A.; Li, F.; Ingram, D.A.; Menick, D.; Field, L.; et al. Periostin Is Required for Maturation and Extracellular Matrix Stabilization of Noncardiomyocyte Lineages of the Heart. Circ. Res. 2008, 102, 752–760. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W., 2nd; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef]
- Shimazaki, M.; Nakamura, K.; Kii, I.; Kashima, T.; Amizuka, N.; Li, M.; Saito, M.; Fukuda, K.; Nishiyama, T.; Kitajima, S.; et al. Periostin is essential for cardiac healingafter acute myocardial infarction. J. Exp. Med. 2008, 205, 295–303. [Google Scholar] [CrossRef]
- Fenderson, B.A.; Stamenkovic, I.; Aruffo, A. Localization of hyaluronan in mouse embryos during implantation, gastrulation and organogenesis. Differentiation 1993, 54, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Waldenström, A.; Martinussen, H.J.; Gerdin, B.; Hällgren, R. Accumulation of hyaluronan and tissue edema in experimental myocardial infarction. J. Clin. Investig. 1991, 88, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Domenech, M.; Polo-Corrales, L.; Ramirez-Vick, J.E.; Freytes, D.O. Tissue Engineering Strategies for Myocardial Regeneration: Acellular Versus Cellular Scaffolds? Tissue Eng. Part B Rev. 2016, 22, 438–458. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Urodeles | Teleost | Postnatal Mammals | Adult Mammals | |
|---|---|---|---|---|
| Collagen 1 | Constituent of the permanent scar formed after macrophage ablation [47]. | Increased expression in cardiac fibroblasts after injury [56]. | Less basal content than in adult hearts [106]. | More expression than in the postnatal heart [106]. Constituent of the scar after injury [116]. |
| Collagen 3 | Present [45]. | Not detected [106]. | Present [106]. | |
| Collagen 5 | Increased RNA expression after injury [68]. | Not detected [106]. | Present [106]. | |
| Collagen 7 | Present after resolution of transient [68]. | |||
| Collagen 8 | Expressed by fibroblasts once the transient scar is resolved [56]. | Present. Induces fibrosis and myofibroblast activation in pressure overload models [117]. | ||
| Collagen 10 | Expressed by fibroblasts once the transient scar is resolved [56]. | |||
| Collagen11A1 Collagen11A2 | Expressed by activated fibroblasts after injury [56]. Expressed by fibroblasts once the transient scar is resolved [56]. | |||
| Collagen 12 | Expressed by activated fibroblasts after injury [118]. | |||
| Collagen 13 | Expressed by fibroblasts once the transient scar is resolved [56]. | |||
| Fibronectin | Overexpressed 21 days after injury (before cardiomyocyte migration) [44]. | Expressed in basal conditions [68]. Highly upregulated after injury, prior to cardiomyocyte migration [69]. Ablation induces scar formation and blocks regeneration [69]. | Expressed in basal conditions [106]. | Reduced expression in basal conditions [106]. Specific ablation in fibroblasts reduces fibrosis [109]. |
| Tenascin C | No basal expression but highly expressed 3 days after injury [44]. Reduced expression in scar after macrophage ablation [45]. | Not expressed in normal conditions. Induces fibrosis after injury promoting myofibroblast migration [107]. | ||
| Periostin | Expressed in cardiac fibroblasts after injury [56]. | Expressed in basal conditions [106,119]. | Not expressed in basal conditions [106]. Required for cardiac fibroblast activation and scar formation after injury [23,120,121]. | |
| Hyaluronic acid | Highly expressed 3 days after injury [44]. | Expressed during embryogenesis [122]. | Expressed in basal conditions [122]. Actively formed after injury [123]. | |
| Agrin | Expressed in basal conditions [112]. | Not expressed in basal conditions [112]. | ||
| Fibrillin-2 | Expressed in basal conditions [106]. | Not expressed in basal conditions [106]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hortells, L.; Johansen, A.K.Z.; Yutzey, K.E. Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts. J. Cardiovasc. Dev. Dis. 2019, 6, 29. https://doi.org/10.3390/jcdd6030029
Hortells L, Johansen AKZ, Yutzey KE. Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts. Journal of Cardiovascular Development and Disease. 2019; 6(3):29. https://doi.org/10.3390/jcdd6030029
Chicago/Turabian StyleHortells, Luis, Anne Katrine Z. Johansen, and Katherine E. Yutzey. 2019. "Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts" Journal of Cardiovascular Development and Disease 6, no. 3: 29. https://doi.org/10.3390/jcdd6030029
APA StyleHortells, L., Johansen, A. K. Z., & Yutzey, K. E. (2019). Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts. Journal of Cardiovascular Development and Disease, 6(3), 29. https://doi.org/10.3390/jcdd6030029