Gene Therapy Approaches to Biological Pacemakers



Abstract
1. Introduction
2. Prerequisites for the Generation of a Biological Pacemaker
3. Cell-Based Approaches to Biological Pacemakers
4. General Principles of Myocardial Gene Therapy
4.1. Vectors
Improving rAAV Delivery Vectors
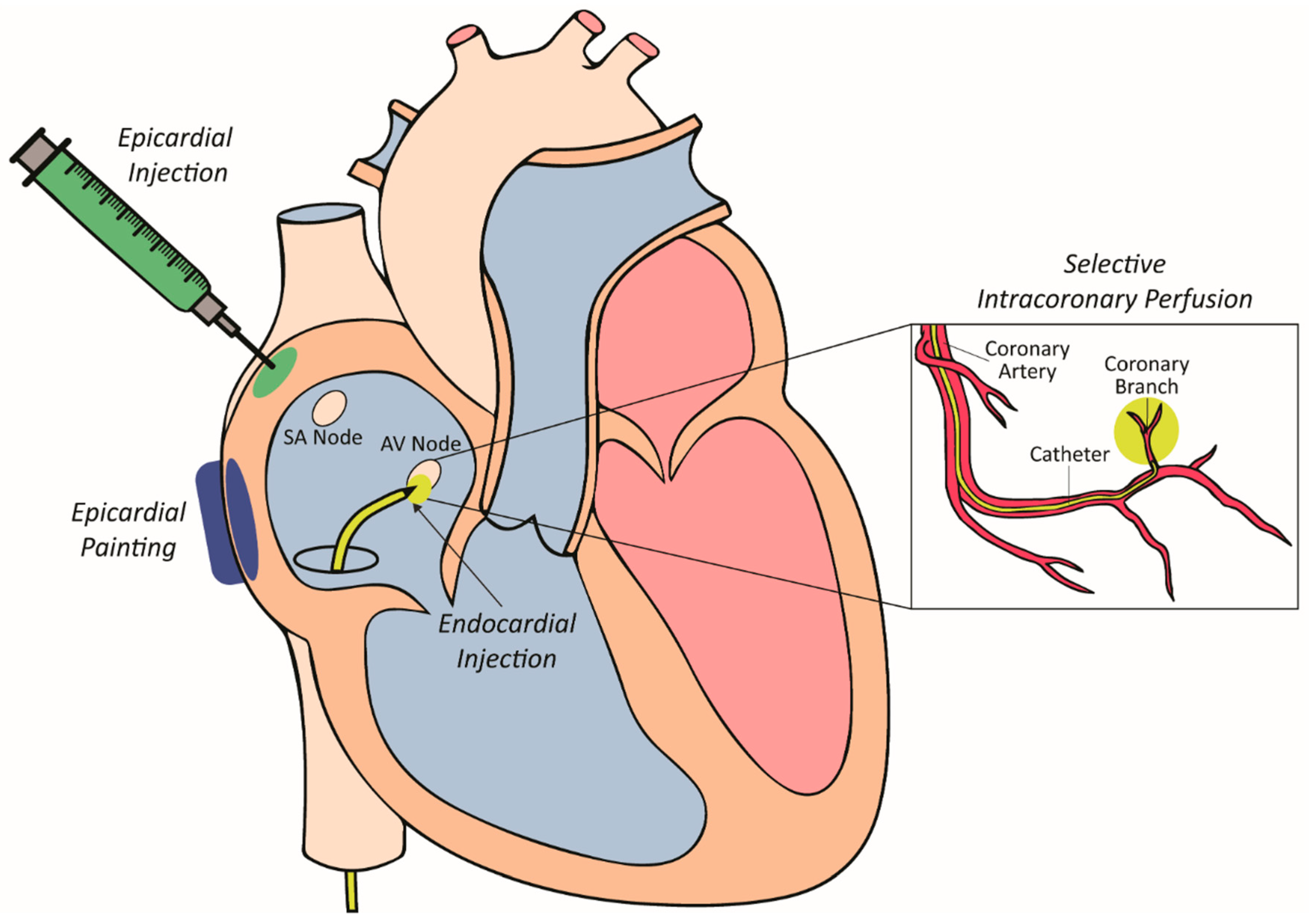
4.2. Vector Delivery to Myocardium
4.2.1. Epicardial Painting
4.2.2. Epicardial Injection
4.2.3. Selective Intracoronary Perfusion
4.2.4. Intramyocardial Injection
5. Gene Therapy Approaches to Biological Pacemakers
5.1. Receptor-Based Gene Therapy Apporach
5.2. Channel-Based Gene Therapy Approaches
5.2.1. Kir2.1 Channel Downregulation (Kir2.1AAA)
5.2.2. HCN Overexpression
5.3. Combined Gene-Cell Approaches
5.4. Somatic Reprogramming to an Induced Pacemaker-Like Phenotype
6. RNA and Small Molecule-Based Therapy Approaches
7. Limitations of Gene Therapy Use for Biological Pacemakers
8. Conclusions
Funding
Conflicts of Interest
References
- Bleeker, W.K.; Mackaay, A.J.; Masson-Pévet, M.; Bouman, L.N.; Becker, A.E. Functional and morphological organization of the rabbit sinus node. Circ. Res. 1980, 46, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.; Liang, W.; Marbán, E.; Cho, H.C. Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat. Biotechnol. 2012, 31, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Gregoratos, G. Sick Sinus Syndrome. Circulation 2003, 108, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Semelka, M.; Gera, J.; Usman, S. Sick sinus syndrome: A review. Am. Fam. Physician 2013, 87, 691–696. [Google Scholar] [PubMed]
- Jensen, P.N.; Gronroos, N.N.; Chen, L.Y.; Folsom, A.R.; deFilippi, C.; Heckbert, S.R.; Alonso, A. Incidence of and risk factors for sick sinus syndrome in the general population. J. Am. Coll. Cardiol. 2014, 64, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Adan, V.; Crown, L.A. Diagnosis and treatment of sick sinus syndrome. Am. Fam. Physician 2003, 67, 1725–1732. [Google Scholar] [PubMed]
- Bradshaw, P.J.; Stobie, P.; Knuiman, M.W.; Briffa, T.G.; Hobbs, M.S. Trends in the incidence and prevalence of cardiac pacemaker insertions in an ageing population. Open Heart 2014, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- John, R.M.; Kumar, S. Sinus Node and Atrial Arrhythmias. Circulation 2016, 133, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.B.; Lemberg, L. The Sick Sinus Syndrome. Am. J. Crit. Care 2006, 15, 226–229. [Google Scholar] [PubMed]
- Cronin, B.; Essandoh, M.K. Update on Cardiovascular Implantable Electronic Devices for Anesthesiologists. J. Cardiothorac. Vasc. Anesth. 2017, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Mulpuru, S.K.; Madhavan, M.; McLeod, C.J.; Cha, Y.-M.; Friedman, P.A. Cardiac Pacemakers: Function, Troubleshooting, and Management: Part 1 of a 2-Part Series. J. Am. Coll. Cardiol. 2017, 69, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, M.; Mulpuru, S.K.; McLeod, C.J.; Cha, Y.-M.; Friedman, P.A. Advances and Future Directions in Cardiac Pacemakers: Part 2 of a 2-Part Series. J. Am. Coll. Cardiol. 2017, 69, 211–235. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, E.; Goldhaber, J.I.; Marban, E. Next-generation pacemakers: From small devices to biological pacemakers. Nat. Rev. Cardiol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Mond, H.G.; Freitag, G. The cardiac implantable electronic device power source: Evolution and revolution. Pacing Clin. Electrophysiol. 2014, 37, 1728–1745. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.M.; Purerfellner, H.; Israel, C.W.; Sunthorn, H.; Kacet, S.; Anelli-Monti, M.; Tang, F.; Young, M.; Boriani, G. Reducing unnecessary right ventricular pacing with the managed ventricular pacing mode in patients with sinus node disease and AV block. Pacing Clin. Electrophysiol. 2006, 29, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Banaszewski, M.; Stępińska, J. Right heart perforation by pacemaker leads. Arch. Med. Sci. 2012, 8, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.N.; Joseph, G.; Khaykin, Y.; Ziada, K.M.; Wilkoff, B.L. Delayed Lead Perforation: A Disturbing Trend. Pacing Clin. Electrophysiol. 2005, 28, 251–253. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Borer, J.S. The funny current: Cellular basis for the control of heart rate. Drugs 2007, 67 (Suppl. 2), 15–24. [Google Scholar] [CrossRef]
- Dobrzynski, H.; Boyett, M.R.; Anderson, R.H. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation 2007, 115, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Baruscotti, M.; Bucchi, A.; Difrancesco, D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Miake, J.; Marban, E.; Nuss, H.B. Biological pacemaker created by gene transfer. Nature 2002, 419, 132–133. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Schulze–Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakamura, K.; Hayashi, T.; Inagaki, N.; Takahashi, M.; Arimura, T.; Morita, H.; Higashiuesato, Y.; Hirano, Y.; Yasunami, M.; et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 2004, 279, 27194–27198. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, K.; Yoshida, M.; Sugiyama, H.; Ohe, T.; Kurokawa, J.; Furukawa, T.; Takano, M.; Nagase, S.; Morita, H.; et al. Enhancement of Spontaneous Activity by HCN4 Overexpression in Mouse Embryonic Stem Cell-Derived Cardiomyocytes—A Possible Biological Pacemaker. PLoS ONE 2015, 10, e0138193. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, K.; Yoshida, M.; Sugiyama, H.; Takano, M.; Nagase, S.; Morita, H.; Kusano, K.F.; Ito, H. HCN4-Overexpressing Mouse Embryonic Stem Cell-Derived Cardiomyocytes Generate a New Rapid Rhythm in Rats with Bradycardia. Int. Heart J. 2018, 59, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, R.; Markandeya, Y.S.; Kamp, T.J.; Makielski, J.C.; January, C.T.; Eckhardt, L.L. IK1-enhanced human-induced pluripotent stem cell-derived cardiomyocytes: An improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am. J. Physiol. Heart. Circ. Physiol. 2016, 310, H1611–H1621. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.-F.; Siu, C.-W.; Chan, Y.-C.; Lau, Y.-M.; Lau, C.-P.; Li, R.A. Synergistic effects of Inward Rectifier (IK1) and Pacemaker (If) Currents on the Induction of Bioengineered Cardiac Automaticity. J. Cardiovasc. Electrophysiol. 2009, 20, 1048–1054. [Google Scholar]
- Xue, T.; Cho, H.C.; Akar, F.G.; Tsang, S.Y.; Jones, S.P.; Marban, E.; Tomaselli, G.F.; Li, R.A. Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: Insights into the development of cell-based pacemakers. Circulation 2005, 111, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ionta, V.; Liang, W.; Kim, E.H.; Rafie, R.; Giacomello, A.; Marbán, E.; Cho, H.C. SHOX2 overexpression favors differentiation of embryonic stem cells into cardiac pacemaker cells, improving biological pacing ability. Stem Cell Rep. 2015, 4, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, K.; Ito, H. Cell-based Biological Pacemakers: Progress and Problems. Acta Med. Okayama 2018, 72, 1–7. [Google Scholar] [PubMed]
- Gepstein, L. Stem cells as biological heart pacemakers. Expert Opin. Biol. Ther. 2005, 5, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Ruhparwar, A.; Tebbenjohanns, J.; Niehaus, M.; Mengel, M.; Irtel, T.; Kofidis, T.; Pichlmaier, A.M.; Haverich, A. Transplanted fetal cardiomyocytes as cardiac pacemaker. Eur. J. Cardiothorac. Surg. 2002, 21, 853–857. [Google Scholar] [CrossRef]
- Xu, C.; Police, S.; Rao, N.; Carpenter, M.K. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ. Res. 2002, 91, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Kehat, I.; Khimovich, L.; Caspi, O.; Gepstein, A.; Shofti, R.; Arbel, G.; Huber, I.; Satin, J.; Itskovitz-Eldor, J.; Gepstein, L. Electromechanical integration of cardiomyocytes derived from human embryonic stem cells. Nat. Biotechnol. 2004, 22, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Mandel, Y.; Weissman, A.; Schick, R.; Barad, L.; Novak, A.; Meiry, G.; Goldberg, S.; Lorber, A.; Rosen, M.R.; Itskovitz-Eldor, J.; et al. Human Embryonic and Induced Pluripotent Stem Cell–Derived Cardiomyocytes Exhibit Beat Rate Variability and Power-Law Behavior. Circulation 2012, 125, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, S.; Anyukhovskiy, Y.; Benari, M.; Naor, S.; Danilo, P.; Rahim, T.; Potapova, I.; Burke, S.; Jiang, Y.-P.; Qiu, X.; et al. Keratinocyte-derived cardiomyocytes provide in vivo biological pacemaker function. Arch. Cardiovasc. Dis. Suppl. 2016, 8, 257. [Google Scholar]
- Jung, J.J.; Husse, B.; Rimmbach, C.; Krebs, S.; Stieber, J.; Steinhoff, G.; Dendorfer, A.; Franz, W.-M.; David, R. Programming and Isolation of Highly Pure Physiologically and Pharmacologically Functional Sinus-Nodal Bodies from Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, S.; Anyukhovsky, E.P.; Ben-Ari, M.; Naor, S.; Jiang, Y.P.; Danilo, P., Jr.; Rahim, T.; Burke, S.; Qiu, X.; Potapova, I.A.; et al. Induced Pluripotent Stem Cell-Derived Cardiomyocytes Provide in vivo Biological Pacemaker Function. Circ. Arrhythm. Electrophysiol. 2017, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Masson-Pevet, M.A.; Bleeker, W.K.; Besselsen, E.; Treytel, B.W.; Jongsma, H.J.; Bouman, L.N. Pacemaker cell types in the rabbit sinus node: A correlative ultrastructural and electrophysiological study. J. Mol. Cell. Cardiol. 1984, 16, 53–63. [Google Scholar] [CrossRef]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Dobrzynski, H.; Li, J.; Tellez, J.; Greener, I.D.; Nikolski, V.P.; Wright, S.E.; Parson, S.H.; Jones, S.A.; Lancaster, M.K.; Yamamoto, M.; et al. Computer three-dimensional reconstruction of the sinoatrial node. Circulation 2005, 111, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, Y.; Lü, S.; Zhou, J.; Du, Z.; Duan, C.; Li, Z.; Wang, C. The tumourigenicity of iPS cells and their differentiated derivates. J. Cell. Mol. Med. 2013, 17, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.; Narsinh, K.H.; Wu, J.C. Derivation of human induced pluripotent stem cells for cardiovascular disease modeling. Circ. Res. 2011, 108, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Lugenbiel, P.; Thomas, D.; Kelemen, K.; Trappe, K.; Bikou, O.; Schweizer, P.A.; Voss, F.; Becker, R.; Katus, H.A.; Bauer, A. Genetic suppression of Galphas protein provides rate control in atrial fibrillation. Basic Res. Cardiol. 2012, 107, 265. [Google Scholar] [CrossRef] [PubMed]
- Trappe, K.; Thomas, D.; Bikou, O.; Kelemen, K.; Lugenbiel, P.; Voss, F.; Becker, R.; Katus, H.A.; Bauer, A. Suppression of persistent atrial fibrillation by genetic knockdown of caspase 3: A pre-clinical pilot study. Eur. Heart J. 2013, 34, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Gersbach, C.A. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [PubMed]
- French, B.A.; Mazur, W.; Geske, R.S.; Bolli, R. Direct in vivo gene transfer into porcine myocardium using replication-deficient adenoviral vectors. Circulation 1994, 90, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Kizana, E.; Alexander, I.E. Cardiac gene therapy: Therapeutic potential and current progress. Curr. Gene. Ther. 2003, 3, 418–451. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.Y.; VandenDriessche, T.; Chuah, M.K. Gene therapy for cardiovascular disease: Advances in vector development, targeting, and delivery for clinical translation. Cardiovasc. Res. 2015, 108, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Wolf, G.D.; Schmidt-Wolf, I.G.H. Non-viral and hybrid vectors in human gene therapy: An update. Trends Mol. Med. 2003, 9, 67–72. [Google Scholar] [CrossRef]
- Liu, Z.; Donahue, J.K. The Use of Gene Therapy for Ablation of Atrial Fibrillation. Arrhythm. Electrophysiol. Rev. 2014, 3, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zaiss, A.K.; Colarusso, P.; Patel, K.; Haljan, G.; Wickham, T.J.; Muruve, D.A. The role of capsid-endothelial interactions in the innate immune response to adenovirus vectors. Hum. Gene Ther. 2003, 14, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A. The innate immune response to adenovirus vectors. Hum. Gene Ther. 2004, 15, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Lusky, M.; Christ, M.; Rittner, K.; Dieterle, A.; Dreyer, D.; Mourot, B.; Schultz, H.; Stoeckel, F.; Pavirani, A.; Mehtali, M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998, 72, 2022–2032. [Google Scholar] [PubMed]
- Gorziglia, M.I.; Kadan, M.J.; Yei, S.; Lim, J.; Lee, G.M.; Luthra, R.; Trapnell, B.C. Elimination of both E1 and E2 from adenovirus vectors further improves prospects for in vivo human gene therapy. J. Virol. 1996, 70, 4173–4178. [Google Scholar] [PubMed]
- Zhao, J.; Pettigrew, G.J.; Thomas, J.; Vandenberg, J.I.; Delriviere, L.; Bolton, E.M.; Carmichael, A.; Martin, J.L.; Marber, M.S.; Lever, A.M.L. Lentiviral vectors for delivery of genes into neonatal and adult ventricular cardiac myocytes in vitro and in vivo. Basic Res. Cardiol. 2002, 97, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.D.; Ranjzad, P.; Kakar, S.J.; Kingston, P.A. Development of Viral Vectors for Use in Cardiovascular Gene Therapy. Viruses 2010, 2, 334–371. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.; Riyad, J.M.; Weber, T. Cardiac gene therapy with adeno-associated virus-based vectors. Curr. Opin. Cardiol. 2017, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Zentilin, L.; Giacca, M. Adeno-associated virus vectors as therapeutic and investigational tools in the cardiovascular system. Circ. Res. 2014, 114, 1827–1846. [Google Scholar] [CrossRef] [PubMed]
- Pacak, C.A.; Byrne, B.J. AAV vectors for cardiac gene transfer: Experimental tools and clinical opportunities. Mol. Ther. 2011, 19, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Farraha, M.; Chong, J.J.; Kizana, E. Therapeutic Prospects of Gene Therapy for Atrial Fibrillation. Heart Lung Circ. 2016, 25, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Asokan, A.; Conway, J.C.; Phillips, J.L.; Li, C.; Hegge, J.; Sinnott, R.; Yadav, S.; DiPrimio, N.; Nam, H.J.; Agbandje-McKenna, M.; et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010, 28, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, L.; Johnson, J.S.; Zhijian, W.; Grieger, J.C.; Ping-Jie, X.; Drouin, L.M.; Agbandje-McKenna, M.; Pickles, R.J.; Samulski, R.J. Generation of Novel AAV Variants by Directed Evolution for Improved CFTR Delivery to Human Ciliated Airway Epithelium. Mol. Ther. 2009, 17, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Paulk, N.K.; Pekrun, K.; Lisowski, L.; Zhang, Y.; Chu, K.; Kay, M.A. Directed Evolution of Improved AAV Capsids for the Ideal Human Liver Vector, 2013; Can Human Liver Tropism and Human Immune Evasion Be Achieved? Mol. Ther. 2015, 23, S104–S105. [Google Scholar] [CrossRef]
- Yang, L.; Li, J.; Xiao, X. Directed evolution of adeno-associated virus (AAV) as vector for muscle gene therapy. Methods Mol. Biol. 2011, 709, 127–139. [Google Scholar] [PubMed]
- Hu, Y.F.; Dawkins, J.F.; Cho, H.C.; Marban, E.; Cingolani, E. Biological pacemaker created by minimally invasive somatic reprogramming in pigs with complete heart block. Sci. Transl. Med. 2014, 6, 245–294. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.N.; Sosunov, E.A.; Qu, J.; Shlapakova, I.N.; Anyukhovsky, E.P.; Liu, L.; Janse, M.J.; Brink, P.R.; Cohen, I.S.; Robinson, R.B.; et al. Biological pacemaker implanted in canine left bundle branch provides ventricular escape rhythms that have physiologically acceptable rates. Circulation 2004, 109, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; McDonald, A.D.; Sasano, T.; Donahue, J.K. Targeted modification of atrial electrophysiology by homogeneous transmural atrial gene transfer. Circulation 2005, 111, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Finet, J.E.; Takeuchi, A.; Fujino, Y.; Strom, M.; Greener, I.D.; Rosenbaum, D.S.; Donahue, J.K. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation 2012, 125, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.K. Biological Therapies for Atrial Fibrillation: Ready for Prime Time? J. Cardiovasc. Pharmacol. 2016, 67, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Boink, G.J.; Duan, L.; Nearing, B.D.; Shlapakova, I.N.; Sosunov, E.A.; Anyukhovsky, E.P.; Bobkov, E.; Kryukova, Y.; Ozgen, N.; Danilo, P., Jr.; et al. HCN2/SkM1 gene transfer into canine left bundle branch induces stable, autonomically responsive biological pacing at physiological heart rates. J. Am. Coll. Cardiol. 2013, 61, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Aistrup, G.L.; Cokic, I.; Ng, J.; Gordon, D.; Koduri, H.; Browne, S.; Arapi, D.; Segon, Y.; Goldstein, J.; Angulo, A.; et al. Targeted nonviral gene-based inhibition of Galpha(i/o)-mediated vagal signaling in the posterior left atrium decreases vagal-induced atrial fibrillation. Heart Rhythm 2011, 8, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Bikou, O.; Thomas, D.; Trappe, K.; Lugenbiel, P.; Kelemen, K.; Koch, M.; Soucek, R.; Voss, F.; Becker, R.; Katus, H.A.; et al. Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model. Cardiovasc. Res. 2011, 92, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Ueno, H.; Pan, Y.; Tomita, H.; Yamamoto, H.; Kanegae, Y.; Saito, I.; Takeshita, A. Percutaneous transluminal gene transfer into canine myocardium in vivo by replication-defective adenovirus. Cardiovasc. Res. 1995, 30, 97–105. [Google Scholar] [CrossRef]
- Hargrave, B.; Downey, H.; Strange, R., Jr.; Murray, L.; Cinnamond, C.; Lundberg, C.; Israel, A.; Chen, Y.J.; Marshall, W., Jr.; Heller, R. Electroporation-mediated gene transfer directly to the swine heart. Gene. Ther. 2013, 20, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.K.; Heldman, A.W.; Fraser, H.; McDonald, A.D.; Miller, J.M.; Rade, J.J.; Eschenhagen, T.; Marban, E. Focal modification of electrical conduction in the heart by viral gene transfer. Nat. Med. 2000, 6, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.K.; Kikkawa, K.; Thomas, A.D.; Marban, E.; Lawrence, J.H. Acceleration of widespread adenoviral gene transfer to intact rabbit hearts by coronary perfusion with low calcium and serotonin. Gene Ther. 1998, 5, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Hadri, L.; Rapti, K.; Ladage, D.; Ishikawa, K.; Tilemann, L.; Yi, G.H.; Morel, C.; Gwathmey, J.K.; Zsebo, K.; et al. Concomitant intravenous nitroglycerin with intracoronary delivery of AAV1.SERCA2a enhances gene transfer in porcine hearts. Mol. Ther. 2012, 20, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.M.; Lai, N.C.; Gao, M.H.; Fine, S.; McKirnan, M.D.; Roth, D.A.; Hammond, H.K. Nitroprusside increases gene transfer associated with intracoronary delivery of adenovirus. Hum. Gene Ther. 2004, 15, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Marban, E.; Lawrence, J.H.; Donahue, J.K. Phosphodiesterase inhibitor-mediated potentiation of adenovirus delivery to myocardium. J. Mol. Cell. Cardiol. 2001, 33, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Sasano, T.; Kikuchi, K.; McDonald, A.D.; Lai, S.; Donahue, J.K. Targeted High-Efficiency, Homogeneous Myocardial Gene Transfer. J. Mol. Cell. Cardiol. 2007, 42, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.K.; Kikkawa, K.; Johns, D.C.; Marban, E.; Lawrence, J.H. Ultrarapid, highly efficient viral gene transfer to the heart. Proc. Natl. Acad. Sci. USA 1997, 94, 4664–4668. [Google Scholar] [CrossRef] [PubMed]
- Lugenbiel, P.; Bauer, A.; Kelemen, K.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. Biological Heart Rate Reduction Through Genetic Suppression of Gαs Protein in the Sinoatrial Node. J. Am. Heart Assoc. 2012, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Edelberg, J.M.; Aird, W.C.; Rosenberg, R.D. Enhancement of murine cardiac chronotropy by the molecular transfer of the human beta2 adrenergic receptor Cdna. J. Clin. Investig. 1998, 101, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rodefeld, M.D.; Beau, S.L.; Schuessler, R.B.; Boineau, J.P.; Saffitz, J.E. Beta-adrenergic and muscarinic cholinergic receptor densities in the human sinoatrial node: Identification of a high beta 2-adrenergic receptor density. J. Cardiovasc. Electrophysiol. 1996, 7, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; DiFrancesco, D. What keeps us ticking: A funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 2009, 47, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Piron, J.; Quang, K.L.; Briec, F.; Amirault, J.C.; Leoni, A.L.; Desigaux, L.; Escande, D.; Pitard, B.; Charpentier, F. Biological pacemaker engineered by nonviral gene transfer in a mouse model of complete atrioventricular block. Mol. Ther. 2008, 16, 1937–1943. [Google Scholar] [CrossRef] [PubMed]
- Edelberg, J.; Huang, D.; Josephson, M.; Rosenberg, R. Molecular enhancement of porcine cardiac chronotropy. Heart 2001, 86, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Plotnikov, A.N.; Danilo, P., Jr.; Shlapakova, I.; Cohen, I.S.; Robinson, R.B.; Rosen, M.R. Expression and function of a biological pacemaker in canine heart. Circulation 2003, 107, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Miake, J.; Marban, E.; Nuss, H.B. Functional role of inward rectifier current in heart probed by Kir2.1 overexpression and dominant-negative suppression. J. Clin. Investig. 2003, 111, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 1993, 55, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Schneider, A.; Wahl, C. Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc. Med. 2002, 12, 206–212. [Google Scholar] [CrossRef]
- Joung, B.; Tang, L.; Maruyama, M.; Han, S.; Chen, Z.; Stucky, M.; Jones, L.R.; Fishbein, M.C.; Weiss, J.N.; Chen, P.S.; et al. Intracellular calcium dynamics and acceleration of sinus rhythm by beta-adrenergic stimulation. Circulation 2009, 119, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.S.; Robinson, R.B. Pacemaker current and automatic rhythms: Toward a molecular understanding. In Basis and Treatment of Cardiac Arrhythmias; Springer: Berlin/Heidelberg, Germany, 2006; pp. 41–71. [Google Scholar]
- Moosmang, S.; Stieber, J.; Zong, X.; Biel, M.; Hofmann, F.; Ludwig, A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur. J. Biochem. 2001, 268, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Bucchi, A.; Plotnikov, A.N.; Shlapakova, I.; Danilo, P., Jr.; Kryukova, Y.; Qu, J.; Lu, Z.; Liu, H.; Pan, Z.; Potapova, I.; et al. Wild-type and mutant HCN channels in a tandem biological-electronic cardiac pacemaker. Circulation 2006, 114, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.F.; Xue, T.; Lau, C.P.; Siu, C.W.; Wang, K.; Zhang, Q.Y.; Tomaselli, G.F.; Akar, F.G.; Li, R.A. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN Channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation 2006, 114, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.N.; Shlapakova, I.; Szabolcs, M.J.; Danilo, P., Jr.; Lorell, B.H.; Potapova, I.A.; Lu, Z.; Rosen, A.B.; Mathias, R.T.; Brink, P.R.; et al. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation 2007, 116, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Potapova, I.; Plotnikov, A.; Lu, Z.; Danilo, P., Jr.; Valiunas, V.; Qu, J.; Doronin, S.; Zuckerman, J.; Shlapakova, I.N.; Gao, J.; et al. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers. Circ. Res. 2004, 94, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Liechty, K.W.; MacKenzie, T.C.; Shaaban, A.F.; Radu, A.; Moseley, A.M.; Deans, R.; Marshak, D.R.; Flake, A.W. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat. Med. 2000, 6, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.C.; Kashiwakura, Y.; Marban, E. Creation of a biological pacemaker by cell fusion. Circ. Res. 2007, 100, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Espinoza-Lewis, R.A.; Chen, C.; Hu, X.; Zhang, Y.; Chen, Y. The role of Shox2 in SAN development and function. Pediatr. Cardiol. 2012, 33, 882–889. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Svensson, E.C. Setting the pace: Tbx3 and Tbx18 in cardiac conduction system development. Circ. Res. 2009, 104, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Lieberman, J. Special delivery: Targeted therapy with small RNAs. Gene Ther. 2011, 18, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J. RNA-based Therapeutics: Current Progress and Future Prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Zangi, L.; Lui, K.O.; von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Spater, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Mir, F.F.; Jhunjhunwala, S.; Kaczmarek, J.C.; Hurtado, J.E.; Yang, J.H.; Webber, M.J.; Kowalski, P.S.; Heartlein, M.W.; DeRosa, F.; et al. Efficacy and Immunogenicity of Unmodified and Pseudouridine-Modified mRNA Delivered Systemically with Lipid Nanoparticles in vivo. Biomaterials 2016, 109, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gurevich, V.V. Therapeutic Potential of Small Molecules and Engineered Proteins. Handb. Exp. Pharmacol. 2014, 219, 1–12. [Google Scholar] [PubMed]


{kind=link}
| Vector | Plasmid | AAV | Lentivirus | Adenovirus |
|---|---|---|---|---|
| Maximum titre (particles per mL) | N/A | Up to 1013 | Up to 109 | Up to 1013 |
| Genome/Size (Kb) | DNA | ssDNA | ssRNA | dsDNA |
| Insert capacity | 15 kB | 4.8 kB | 10 kB | 7 to 30 kB |
| Integration | No | No | Yes (Random) | No |
| Length of transgene expression | Up to 2 Months | Long Term | Long Term | Up to 2 Weeks |
| Immunogenicity | Minimally Immunogenic | Minimally Immunogenic | Minimally Immunogenic | Cytotoxic and Immunogenic |
| Limited by neutralizing antibodies | No | Yes | No | No |
| Target cells | Dividing and Non-dividing cells | Dividing and Non-dividing cells | Dividing and Non-dividing cells | Dividing and Non-dividing cells |
| Cardiac gene transfer | Low Cardiac Transduction | Cardiotropic AAV Serotypes | Lower Cardiac Transduction | High Cardiac Transduction |
| Disadvantages | Low Transfection Efficiency | Risk of neutralizing antibodies and T-Cell Responses | Risk of insertional mutagenesis | High antibody and inflammatory response |
| Clinical trial approval | Yes | Yes | Yes | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farraha, M.; Kumar, S.; Chong, J.; Cho, H.C.; Kizana, E. Gene Therapy Approaches to Biological Pacemakers. J. Cardiovasc. Dev. Dis. 2018, 5, 50. https://doi.org/10.3390/jcdd5040050
Farraha M, Kumar S, Chong J, Cho HC, Kizana E. Gene Therapy Approaches to Biological Pacemakers. Journal of Cardiovascular Development and Disease. 2018; 5(4):50. https://doi.org/10.3390/jcdd5040050
Chicago/Turabian StyleFarraha, Melad, Saurabh Kumar, James Chong, Hee Cheol Cho, and Eddy Kizana. 2018. "Gene Therapy Approaches to Biological Pacemakers" Journal of Cardiovascular Development and Disease 5, no. 4: 50. https://doi.org/10.3390/jcdd5040050
APA StyleFarraha, M., Kumar, S., Chong, J., Cho, H. C., & Kizana, E. (2018). Gene Therapy Approaches to Biological Pacemakers. Journal of Cardiovascular Development and Disease, 5(4), 50. https://doi.org/10.3390/jcdd5040050