A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease


Abstract
1. Introduction
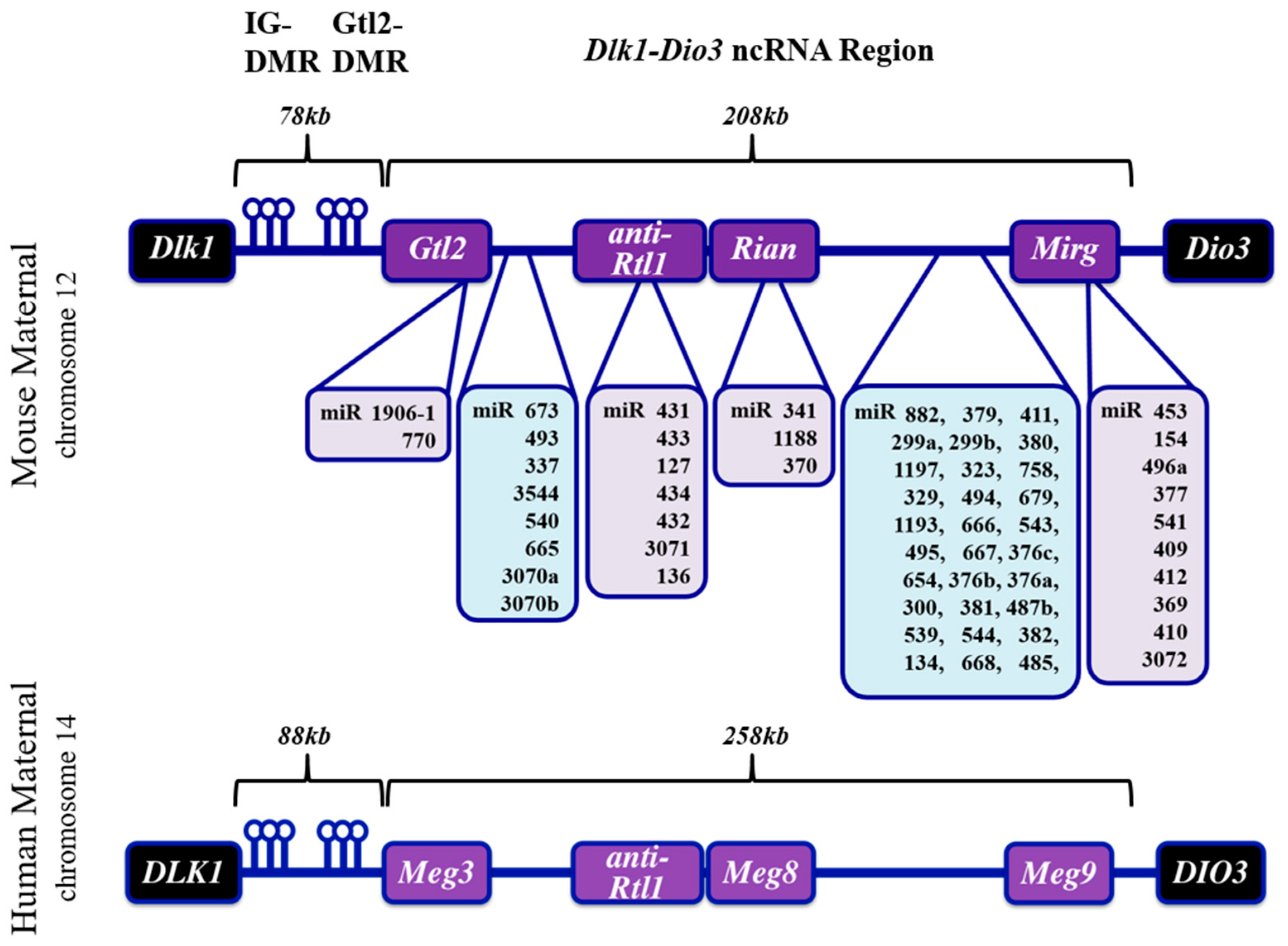
1.1. The Dlk-Dio3 ncRNA Locus: Gene Organization, Imprinting and Human Disease
1.2. The Dlk1-Dio3 ncRNA Locus in Striated Muscle
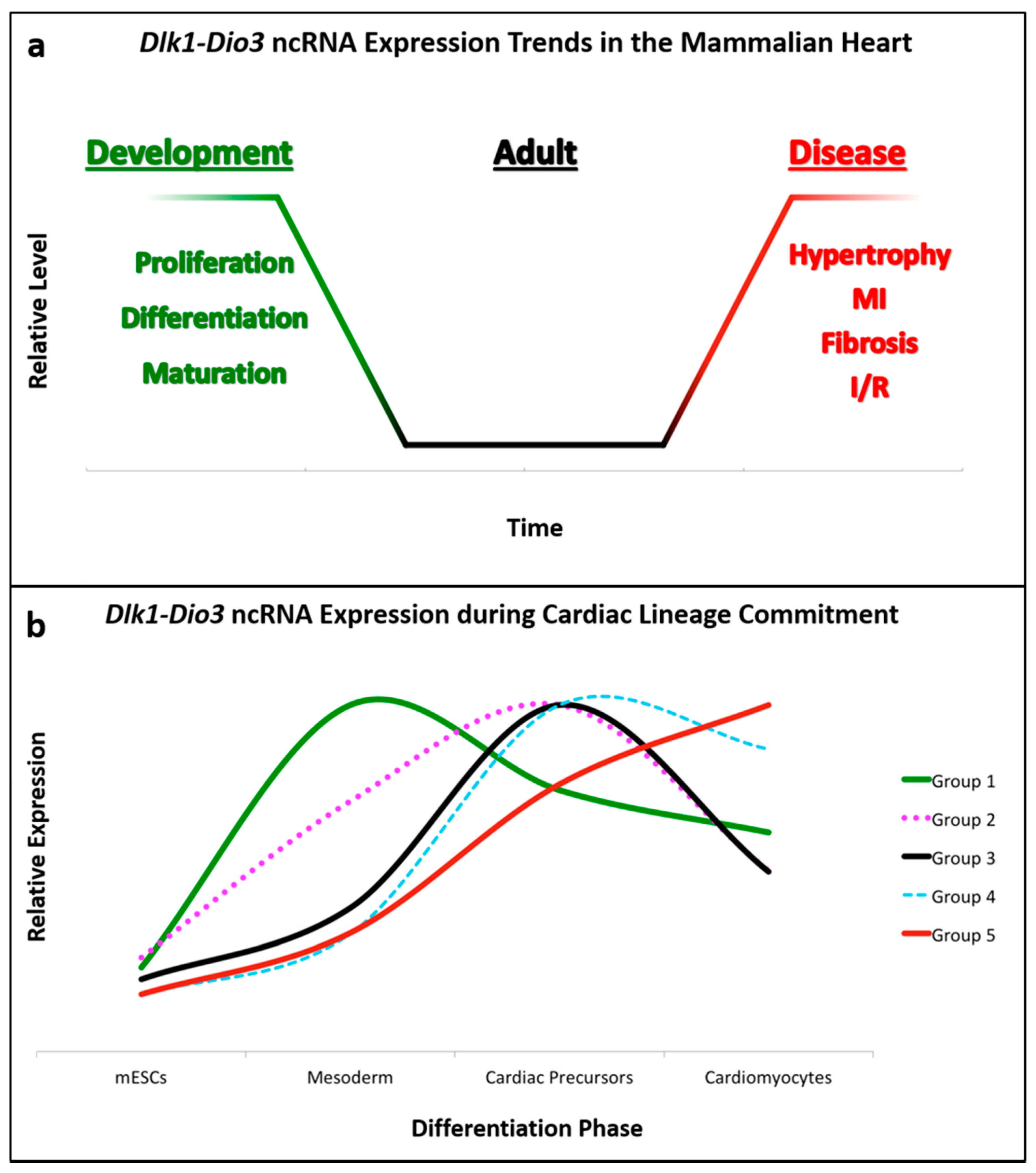
2. MicroRNAs Expressed from the Dlk1-Dio3 Locus in Cardiac Differentiation and Development
2.1. Cardiomyocyte and Endothelial Progenitors
2.2. Cardiac Structures
2.3. Cardiomyocyte Proliferation and Apoptosis
3. MicroRNAs Expressed from the Dlk1-Dio3 Locus in Cardiovascular Pathology
3.1. Cardiac Hypertrophy
3.2. Myocardial Infarction
3.3. Cardiac Fibroblasts
3.4. Ischemia/Reperfusion
3.5. Circulating miRNAs in Cardiovascular Disease
4. Long noncoding RNAs Expressed from the Dlk1-Dio3 Locus in Cardiac Development and Disease
4.1. Gtl2/Meg3 in Cardiomyocytes
4.2. Gtl2/Meg3 in Cardiac Fibroblasts and Pressure Overload
4.3. Gtl2/Meg3 in the Vasculature
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, D.-Z. microRNAs in cardiovascular development. J. Mol. Cell. Cardiol. 2012, 52, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.; Pedrazzini, T.; Chang, C.-P.; Dorn II, G.; Thum, T.; Heymans, S. Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Condorelli, G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat. Rev. Cardiol. 2015, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA Regulatory Networks in Cardiovascular Development. Environ. Health 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Hoelscher, S.C.; Doppler, S.A.; Dreßen, M.; Lahm, H.; Lange, R.; Krane, M. MicroRNAs: Pleiotropic players in congenital heart disease and regeneration. J. Thorac. Dis. 2017, 9, S64–S81. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, J.C.; Boyer, L.A. Getting to the heart of the matter: Long non-coding RNAs in cardiac development and disease. EMBO J. 2013, 32, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.; Tsuchihachi, T.; McManus, M.; Schwartz, R.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Murchison, E.P.; Tang, R.; Callis, T.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.; Schneider, M.; Selzman, C.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Greene, S.B.; Bonilla-claudio, M.; Tao, Y.; Zhang, J.; Bai, Y.; Huang, Z.; Black, B.; Wang, F.; Martin, J. Bmp-signaling regulates myocardial differentiation from cardiac progenitors through a micro RNA-mediated mechanism. Dev. Cell 2010, 19, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-M.; Chen, H.-C.; Chen, S.-J.; Huang, C.-Y.; Wu, T.-W.; Feng, L.-Y.; Tsai, H.-C.; Lui, T.-N.; Hsueh, C.; Wei, K.-C. MicroRNA-495 inhibits proliferation of glioblastoma multiforme cells by downregulating cyclin-dependent kinase 6. World J. Surg. Oncol. 2013, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.; Bassel-Duby, R.; Olson, E. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-P.; Chen, J.-F.; Regan, J.; Maguire, C.; Tang, R.-H.; Dong, X.-R.; Majesky, M.; Wang, D.-Z. Loss of miRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.-J.; Matkovich, S.; Dorn II, G.; van Rooij, E.; Olson, E. The miR-15 Family Regulates Post-natal Mitotic Arrest of Cardiomyocytes Enzo. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.-F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, M.; Naseem, R.; Marshall, W.; Hill, J.; Olson, E. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werbner, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nuernberg, S.; Jin, K.; Xu, W.; Lin, C.-Y.; Lin, C.-J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.-Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-H.; Yuan, Y.-X.; Rao, S.-L.; Wang, P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3653–3660. [Google Scholar] [PubMed]
- Wang, K.; Long, B.; Zhou, L.-Y.; Liu, F.; Zhou, Q.-Y.; Liu, C.-Y.; Fan, Y.-Y.; Li, P.-F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014, 5, 3596. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Royo, H.; Bortolin, M.-L.; Lin, S.-P.; Ferguson-Smith, A.C.; Cavaillé, J. A Large Imprinted microRNA Gene Cluster at the Mouse Dlk1-Gtl2 Domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Hagan, J.P.; O’Neill, B.L.; Stewart, C.L.; Kozlov, S.V.; Croce, C.M. At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-P.; Coan, P.; da Rocha, S.T.; Seitz, H.; Cavaille, J.; Teng, P.-W.; Takada, S.; Ferguson-Smith, A. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development 2007, 134, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.S.; Yevtodiyenko, A.; Schmidt, C.; Schmidt, J.V. Allele-specific histone modifications regulate expression of the Dlk1-Gtl2 imprinted domain. Genomic 2007, 89, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Falix, F.A.; Aronson, D.C.; Lamers, W.H.; Gaemers, I.C. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Dentice, M.; Salvatore, D. Local impact of thyroid hormone inactivation. J. Endocrinol. 2011, 209, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Bonasio, R.; Saldaña-meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Jing, Z.C.; Ellinor, P.T.; Liang, D.; Zhang, H.; Liu, Y.; Chen, X.; Pan, L.; Lyon, R.; Liu, Y.; et al. MicroRNA-134 as a potential plasma biomarker for the diagnosis of acute pulmonary embolism. J. Transl. Med. 2011, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Guntrum, M.; Vlasova, E.; Davis, T.L. Asymmetric DNA methylation of CpG dyads is a feature of secondary DMRs associated with the Dlk1/Gtl2 imprinting cluster in mouse. Epigenet. Chromatin 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Molecular Mechanisms Leading to the Phenotypic Development in Paternal and Maternal Uniparental Disomy for Chromosome 14. Clin. Pediatr. Endocrinol. 2008, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ioannides, Y.; Lokulo-Sodipe, K.; Mackay, D.J.G.; Davies, J.H.; Temple, I.K. Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. J. Med. Genet. 2014, 51, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Kagami-Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 2016, 61, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Brothman, A.R.; Chen, Z.; Bayrak-Toydemir, P.; Longo, N. Paternal uniparental disomy of chromosome 14: Confirmation of a clinically-recognizable phenotype. Am. J. Med. Genet. A 2004, 130A, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Schwartz, S.; McPherson, E. Paternal uniparental isodisomy for chromosome 14 in a patient with a normal 46, XY karyotype. Am. J. Med. Genet. A 2004, 127A, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, P.; Watkins, M.; Surani, M.A.; Ferguson-Smith, A.C. Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 2000, 127, 4719–4728. [Google Scholar] [PubMed]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Kono, T. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.; Zhong, Y.; Rice, K.; Zhang, L.; Zhang, X.; Gordon, F.; Lidov, H.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.; Longmire, T.A.; Shen, S.S.; Bourdon, A.; Sommer, A.; Gadue, P.; Spira, A.; Gouon-Evans, V.; Murphy, G.; Mostoslavsky, G.; et al. Mouse ES and iPS cells can form similar definitive endoderm despite differences in imprinted genes. J. Clin. Investig. 2011, 121, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Valdmanis, P.N.; Roy-chaudhuri, B.; Kim, H.K.; Sayles, L.; Zheng, Y.; Chuang, C.-H.; Caswell, D.; Chu, K.; Winslow, M.; Sweet-Cordero, E.; et al. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene 2015, 34, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Rice, A.L.; Estrella, N.L.; Held, A.; Kandarian, S.C.; Naya, F.J. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development 2013, 140, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.L.; Zhang, S.; Tellam, R.L.; Brooks, D.; McMillen, I.; Porrello, E.; Botting, K. Regulation of microRNA during cardiomyocyte maturation in sheep. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.; Kim, T.-K.; Greenberg, M.; Schratt, G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Wang, L. Transcriptional mechanism for the paired miR-433 and miR-127 genes by nuclear receptors SHP and ERRγ. Nucleic Acids Res. 2008, 36, 5727–5735. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-Q.; Chen, X.; Wang, P.; Lu, L.; Zhao, W.; Chen, C.; Chen, C.-P.; Tao, T.; Sun, J.; Zheng, Y.-Y.; et al. Regulation of DLK1 by the maternally expressed miR-379/miR-544 cluster may underlie callipyge polar overdominance inheritance. Proc. Natl. Acad. Sci. USA 2015, 112, 13627–13632. [Google Scholar] [CrossRef] [PubMed]
- Labialle, S.; Marty, V.; Bortolin-Cavaillé, M.-L.; Hoareau-Osman, M.; Pradere, J.-P.; Valet, P.; Martin, P.; Cavaille, J. The miR-379/miR-410 cluster at the imprinted Dlk 1 -Dio 3 domain controls neonatal metabolic adaptation. EMBO J. 2014, 33, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Wüst, S.; Dröse, S.; Heidler, J.; Wittig, I.; Klockner, I.; Franko, A.; Bonke, E.; Gunther, S.; Garner, U.; Boettger, T.; et al. Metabolic Maturation during Muscle Stem Cell Differentiation Is Achieved by miR-1/133a-Mediated Inhibition of the Dlk1-Dio3 Mega Gene Cluster. Cell Metab. 2018, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Baghdadi, M.B.; Mella, S.; Gayraud-Morel, B.; Marty, V.; Cavaille, J.; Antoniewski, C.; Tajbaksh, S. Small-RNA sequencing identifies dynamic microRNA deregulation during skeletal muscle lineage progression. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Park, Y.-K.; Lee, K.-P.; Lee, S.-M.; Kong, T.-W.; Kim, H.-J.; Dho, S.; Kim, S.-Y.; Kwon, K.-S. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging (Albany NY) 2014, 6, 524–544. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Segers, K.; Karim, L.; Shay, T.; Gyapay, G.; Cockett, N.; Georges, M. The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat. Genet. 2001, 27, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, C.A.; Waddell, J.N.; Taxis, T.M.; Yu, H.; Tellam, R.; Neary, M.; Cockett, N. New insights into polar overdominance in callipyge sheep. Anim. Genet. 2014, 45 (Suppl. 1), 51–61. [Google Scholar] [CrossRef] [PubMed]
- Caiment, F.; Charlier, C.; Hadfield, T.; Cockett, N.; Georges, M.; Baurain, D. Assessing the effect of the CLPG mutation on the microRNA catalogue of skeletal muscle using high throughput sequencing. Genome Res. 2010, 20, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.; Ding, H.; Wylie, J.; Pico, A.; Capra, J.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Tomé, M.; Bernal, J.A.; Bernad, A. MIR-300 mediates Bmi1 function and regulates differentiation in primitive cardiac progenitors. Cell Death Dis. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Zhao, H.; Zhou, L.P.; Zhao, C.-X.; Wu, Y.-F.; Zhen, L.-X.; Li, J.; Ge, D.-X.; Xu, L.; Lin, L.; et al. miR-134 modulates the proliferation of human cardiomyocyte progenitor cells by targeting Meis2. Int. J. Mol. Sci. 2015, 16, 25199–25213. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Huang, W.; Cai, W.; Wang, L.; Guo, L.; Paul, C.; Yu, X.-Y.; Wang, Y. Inhibition of microRNA-495 Enhances Therapeutic Angiogenesis of Human Induced Pluripotent Stem Cells. Stem. Cells 2016, 35, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Vacchi-Suzzi, C.; Hahne, F.; Scheubel, P.; Marcellin, M.; Dubost, V.; Westphal, M.; Boeglen, C.; Buchmann-Moller, S.; Chueng, M.; Cordier, A.; et al. Heart Structure-Specific Transcriptomic Atlas Reveals Conserved microRNA-mRNA Interactions. PLoS ONE 2013, 8, e52442. [Google Scholar] [CrossRef] [PubMed]
- Van der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.J.; Mulder, B.J.M. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2010, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Naya, F.J. MicroRNAs in the myocyte enhancer factor 2 (MEF2)-regulated Gtl2-Dio3 noncoding RNA locus promote cardiomyocyte proliferation by targeting the transcriptional coactivator Cited2. J. Biol. Chem. 2015, 290, 23162–23172. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Zhu, J.G.; Liu, Y.Q. Identification of the microRNA Expression Profile in the Regenerative Neonatal Mouse Heart by Deep Sequencing. Cell Biochem. Biophys. 2014, 70, 635. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Chi, J.Y.; Liang, H.H.; Huangfu, L.-T.; Guo, Z.-D.; Zou, H.; Yin, X.-H. MicroRNA-377 Mediates Cardiomyocyte Apoptosis Induced by Cyclosporin A. Can. J. Cardiol. 2016, 32, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Maruyama, S.; Sano, S.; Accorsi, A.; Girgenrath, M.; Walsh, K.; Naya, F. miR-410 and miR-495 are dynamically regulated in diverse cardiomyopathies and their inhibition attenuates pathological hypertrophy. PLoS ONE 2016, 11, e0151515. [Google Scholar] [CrossRef]
- Weber, K.; Rostert, N.; Bauersachs, S.; Wess, G. Serum microRNA profiles in cats with hypertrophic cardiomyopathy. Mol. Cell. Biochem. 2015, 402, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.M.; Tham, Y.-K.; Ooi, J.; Patterson, N.; Kiriazis, H.; Su, Y.; Thomas, C.; Lin, R.; et al. Inhibition of miR-154 Protects Against Cardiac Dysfunction and Fibrosis in a Mouse Model of Pressure Overload. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ponnusamy, M.; Liu, C.; Tian, J.; Dong, Y.; Gao, J.; Wang, C.; Zhang, Y.; Zhang, L.; Wang, K.; et al. MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, N.; Long, B.; Fan, Y.-Y.; Liu, C.-Y.; Zhou, Q.-Y.; Murtaza, I.; Wang, K.; Li, P.-F. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Zuidwijk, M.; Muller, A.; Mulders, J.; Oudejans, C.B.M.; Simonides, W.S. Cardiac expression of deiodinase type 3 (Dio3) following myocardial infarction is associated with the induction of a pluripotency microRNA signature from the Dlk1-Dio3 genomic region. Endocrinology 2013, 154, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; DeMartino, A.M.; Watson, L.J.; Brittian, K.; Zafir, A.; Dassanyaka, S.; Hong, K.; Jones, S. MicroRNA-539 is up-regulated in failing heart, and suppresses O-GlcNAcase expression. J. Biol. Chem. 2014, 289, 29665–29676. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.; et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, L.; Zhao, J.; Sun, Y.; Yang, H. MiRNA Expression Profile of the Myocardial Tissue of Pigs with Coronary Microembolization. Cell. Physiol. Biochem. 2017, 43, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, N.; Fei, Z.; Qiu, J.; Ma, D.; Liu, X.; Cai, G.; Li, S. Analysis of plasma miR-208a and miR-370 expression levels for early diagnosis of coronary artery disease. Biomed. Rep. 2016, 5, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Y.; Bie, Z.D.; Zhang, C.H.; Li, H.; Li, L.D.; Yang, J. MiR-154 directly suppresses DKK2 to activate Wnt signaling pathway and enhance activation of cardiac fibroblasts. Cell Biol. Int. 2016, 40, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Liu, W.; Wang, D.-Z. MiR-154 promotes myocardial fibrosis through β-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, H.; Jiang, S.; Xu, Y. MicroRNA-495 inhibits the high glucose- induced inflammation, differentiation and extracellular matrix accumulation of cardiac fibroblasts through downregulation of NOD1. Cell. Mol. Biol. Lett. 2018, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Welten, S.M.J.; Bastiaansen, A.J.N.M.; De Jong, R.C.M.; de Vries, M.; Peters, E.; Boonstra, M.; Sheikh, S.; Monica, N.; Kandimalla, E.; Quax, P.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 increases neovascularization and blood flow recovery after ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Joladarashi, D.; Garikipati, V.N.S. Enhanced Cardiac Regenerative Ability of Stem Cells After Ischemia-Reperfusion Injury: Role of Human CD34+ Cells Deficient in MicroRNA-377. J. Am. Coll. Cardiol. 2015, 66, 2214–2226. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Huang, W.; Feng, Y.; Cai, W.; Wang, Y.; Wang, X.; Liang, J.; Wani, M.; Chen, J.; Zhu, P.; et al. MicroRNA-377 regulates mesenchymal stem cell-induced angiogenesis in ischemic hearts by targeting VEGF. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, T.; Dong, Z.; Mi, R. MicroRNA-410 is involved in mitophagy after cardiac ischemia/reperfusion injury by targeting high-mobility group box 1 protein. J. Cell. Biochem. 2018, 119, 2427–2439. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Ren, X.-P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.-C. MicroRNA-494 Targeting both Pro-apoptotic and Anti-apoptotic Proteins Protects against Ischemia/Reperfusion-Induced Cardiac Injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating MicroRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, H.A.; Coskunpinar, E.; Ikitimur, B. The prognostic value of circulating microRNAs in heart failure: Preliminary results from a genome-wide expression study. J. Cardiovasc. Med. 2015, 16, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, J.; Yin, Z.; Wang, F.; Chen, C.; Wang, D.-W. Identification of cardiac-related circulating microRNA profile in human chronic heart failure. Oncotarget 2015, 7, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Möhnle, P.; Schütz, S.V.; Schmidt, M.; Hinske, C.; Hubner, M.; Heyn, J.; Beiras-Fernandez, A.; Kreth, S. MicroRNA-665 is involved in the regulation of the expression of the cardioprotective cannabinoid receptor CB2 in patients with severe heart failure. Biochem. Biophys. Res. Commun. 2014, 451, 516–521. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Lv, P.; Zhao, X.; Wang, X.; Ma, X.; Meng, W.; Meng, X.; Dong, S. Predictive value of circulating miR-328 and miR-134 for acute myocardial infarction. Mol. Cell. Biochem. 2014, 394, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Sakata, Y.; Nakatani, D.; Suna, S.; Mizuno, H.; Shimizu, M.; Usami, M.; Sasaki, T.; Sato, H.; Kawahara, Y.; et al. A subset of circulating microRNAs are predictive for cardiac death after discharge for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2012, 427, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hsiao, Y.-W.; Chang, S.-L. Circulating microRNAs in arrhythmogenic right ventricular cardiomyopathy with ventricular arrhythmia. EP Eur. 2017, 20, f37–f45. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.K.; Loughran, K.A.; Meola, D.M.; Juhr, C.; Thane, K.; Davis, A.; Hoffman, A. Circulating exosome microRNA associated with heart failure secondary to myxomatous mitral valve disease in a naturally occurring canine model. J. Extracell. Vesicles 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Coffey, S.; Williams, M.J.A.; Phillips, L.V.; Jones, G.T. Circulating microRNA Profiling Needs Further Refinement Before Clinical Use in Patients with Aortic Stenosis. J. Am. Heart Assoc. 2015, 4, e002150. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, S.; Huang, D.; Luo, X.; Zhang, J.; Chen, J.; Xu, W. Expression profile analysis of circulating microRNAs and their effects on ion channels in chinese atrial fibrillation patients. Int. J. Clin. Exp. Med. 2015, 8, 845–853. [Google Scholar] [PubMed]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Qiao, Y.; Wang, Q.; Liu, B.; Hou, J.; Li, R.; Tang, C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem. Biophys. Res. Commun. 2018, 495, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nie, X.; Sun, S.; Dong, S.; Yuan, C.; Li, Y.; Xiao, B.; Jie, D.; Liu, Y. Long Non-Coding RNA MEG3 Downregulation Triggers Human Pulmonary Artery Smooth Muscle Cell Proliferation and Migration via the p53 Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Xu, H.; Chang, H.; Tong, Y.; Zhang, T.; Guo, G. Knockdown of long non-coding RNA MEG3 protects H9c2 cells from hypoxia-induced injury by targeting microRNA-183. J. Cell. Biochem. 2018, 119, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Jaé, N.; Holdt, L.; Dimmeler, S. Long Noncoding RNAs from Clinical Genetics to Therapeutic Targets? J. Am. Coll. Cardiol. 2016, 67, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.; Gejman, R.; Ansell, P.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.; Sarma, K.; Song, J.; Kingston, R.; Borrowsky, M.; Lee, J. Genome-wide Identification of Polycomb-Associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Das, P.P.; Hendrix, D.A.; Apostolou, E.; Buchner, A.; Canver, M.; Beyaz, S.; Ljuboja, D.; Kuintzle, R.; Kim, W.; Kamik, R.; et al. PRC2 Is Required to Maintain Expression of the Maternal Gtl2-Rian-Mirg Locus by Preventing De Novo DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep. 2015, 12, 1456–1470. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ncRNA | Development | Species | Disease | Species |
|---|---|---|---|---|
| miRNAs | ||||
| All * | dynamically expressed during mESC cardiomyocyte-directed differentiation [62] | m [62] | ||
| miR-493 | Maturation [51] | s [51] | — | |
| miR-337 | — | MI [77], HF [86], angiogenesis [86], fibrosis [86], remodeling [86]. Targets STK35 [86] | h [86], m [77,86] | |
| miR-665 | — | CHF [92], HF [93] | h [92,93] | |
| miR-431 | Proliferation [15] | m [15], r [15] | — | |
| miR-433 | VSD [68] | h [68] | MI [77,79], fibrosis [79], ventricular remodeling [79]. Targets AZIN1 and JNK1 [79] | m [77,79] |
| miR-127 | Valve morphogenesis [66], maturation [51] | r [66], d [66], mk [66], s [51] | MI [77] | m [77] |
| miR-434 | — | MI [77] | m [77] | |
| miR-432 | Maturation [51] | s [51] | Atrial fibrillation [99] | h [99] |
| miR-136 | — | MI [77] | m [77] | |
| miR-370 | — | MI77, CAD [81], coronary microembolism [80] | m [77], h [81], p [80] | |
| miR-379 | VSD68, maturation [51] | h [68], s [51] | MI [77] | m [77] |
| miR-411 | Proliferation [15], maturation [51] | m [15], r [15], s [51] | MI [77] | m [77] |
| miR-299a | — | Congestive HF [91] | h [91] | |
| miR-299b | — | Congestive HF [91] | h [91] | |
| miR-380 | Proliferation [15], maturation [51] | m [15], r [15], s [51] | MI [95] | h [95] |
| miR-323 | — | Coronary microembolism [80] | p [80] | |
| miR-329 | Maturation [51] | s [51] | MI77, IR-induced neovascularization [85] | m [51,85] |
| miR-494 | — | I/R-induced apoptosis [89], IR-induced neovascularization [85], HF [93], arrhythmogenic RV-cardiomyopathy [96]. Targets PTEN, ROCK1, CamKIIδ, FGFR2, and LIF [89] | m [85,89], h [93,96] | |
| miR-1193 | — | MI [77] | m [77] | |
| miR-543 | Maturation [51] | s [51] | MI [77] | m [77] |
| miR-495 | Proliferation [15,69], angiogenic differentiation [65]. Targets VEZF1 [65], Cited2 [69] | h [65], m [15], r [15,69] | MI neovascularization [65], fibrosis [84], MI [72,77], IR neovascularization [85], mitral valve disease [97] | h [65,84], m [72,77,85], d [97] |
| miR-376c | — | MI [77] | m [77] | |
| miR-654 | VSD [68] | h [68] | — | |
| miR-376b | — | MI [77] | m [77] | |
| miR-376a | — | MI [77] | m [77] | |
| miR-300 | Differentiation [63], target of Bmi1 [63] | m [63] | — | |
| miR-381 | — | Hypertrophy [73] | c [73] | |
| miR-487b | VSD [68], maturation [51] | h [68], s [51] | MI [77], IR neovascularization [85] | m [77,85] |
| miR-539 | Proliferation [15] | m [15], r [15] | MI [77,78], MI apoptosis [28]. Targets OGA [78] and PHB2 [28] | m [28,77,78] |
| miR-544 | — | MI [77] | m [77] | |
| miR-382 | Maturation [51] | s [51] | MI77, comorbid aortic stenosis and CAD [98] | m [77], h [98] |
| miR-134 | Proliferation [64]. Directly targets Meis2 [64] | h [64] | MI [38,94] | h [38,94] |
| miR-668 | Proliferation [15], maturation [70] | m [15,70], r [15] | — | |
| miR-485 | Maturation [51] | s [51] | Hypertrophy [75], MI77, remodeling [75]. Targets MAPL [75] | m [75,77] |
| miR-154 | Proliferation [15], maturation [70] | m [15,70], r [15] | Hypertrophy [74], fibrosis [74,82,83], apoptosis [83]. Targets p15 [74] and DKK2 [82] | m [74,82,83] |
| miR-377 | — | I/R neovascularization [86], MI neovascularization [87], fibrosis [87], Cyclosporin-mediated apoptosis [71]. Targets XIAP [71], NRP2 [71], and VEGF [87] | m [86,87], r [71] | |
| miR-541 | — | Hypertrophy [76], MI 77]. Repressed by MITF [76] | m [76,77] | |
| miR-409 | VSD [68], maturation [51] | h [68], s [51] | MI [77] | m [77] |
| miR-369 | — | MI [77] | m [77] | |
| miR-410 | Proliferation [15,69], maturation [51]. Targets Cited2 [69] | r [15,69], m [15], s [51] | MI [72,77], I/R mitophagy [88]. | m [72,77,88] |
| lncRNAs | ||||
| Gtl2 | Angiogenesis [100,101] | h [100,101] | I/R apoptosis [102], fibrosis [103], hypertension [100], neovascularization [104]. Decoys miR-183 [102] | M [100,103,104], r [102] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dill, T.L.; Naya, F.J. A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2018, 5, 37. https://doi.org/10.3390/jcdd5030037
Dill TL, Naya FJ. A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. Journal of Cardiovascular Development and Disease. 2018; 5(3):37. https://doi.org/10.3390/jcdd5030037
Chicago/Turabian StyleDill, Tiffany L., and Francisco J. Naya. 2018. "A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease" Journal of Cardiovascular Development and Disease 5, no. 3: 37. https://doi.org/10.3390/jcdd5030037
APA StyleDill, T. L., & Naya, F. J. (2018). A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. Journal of Cardiovascular Development and Disease, 5(3), 37. https://doi.org/10.3390/jcdd5030037