ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker

{kind=link}
Abstract
1. Introduction
2. Ex Vivo Evaluation of the ApoB-100 Lipoprotein Proteoglycan Interaction and Its Possible Relation with ACVD
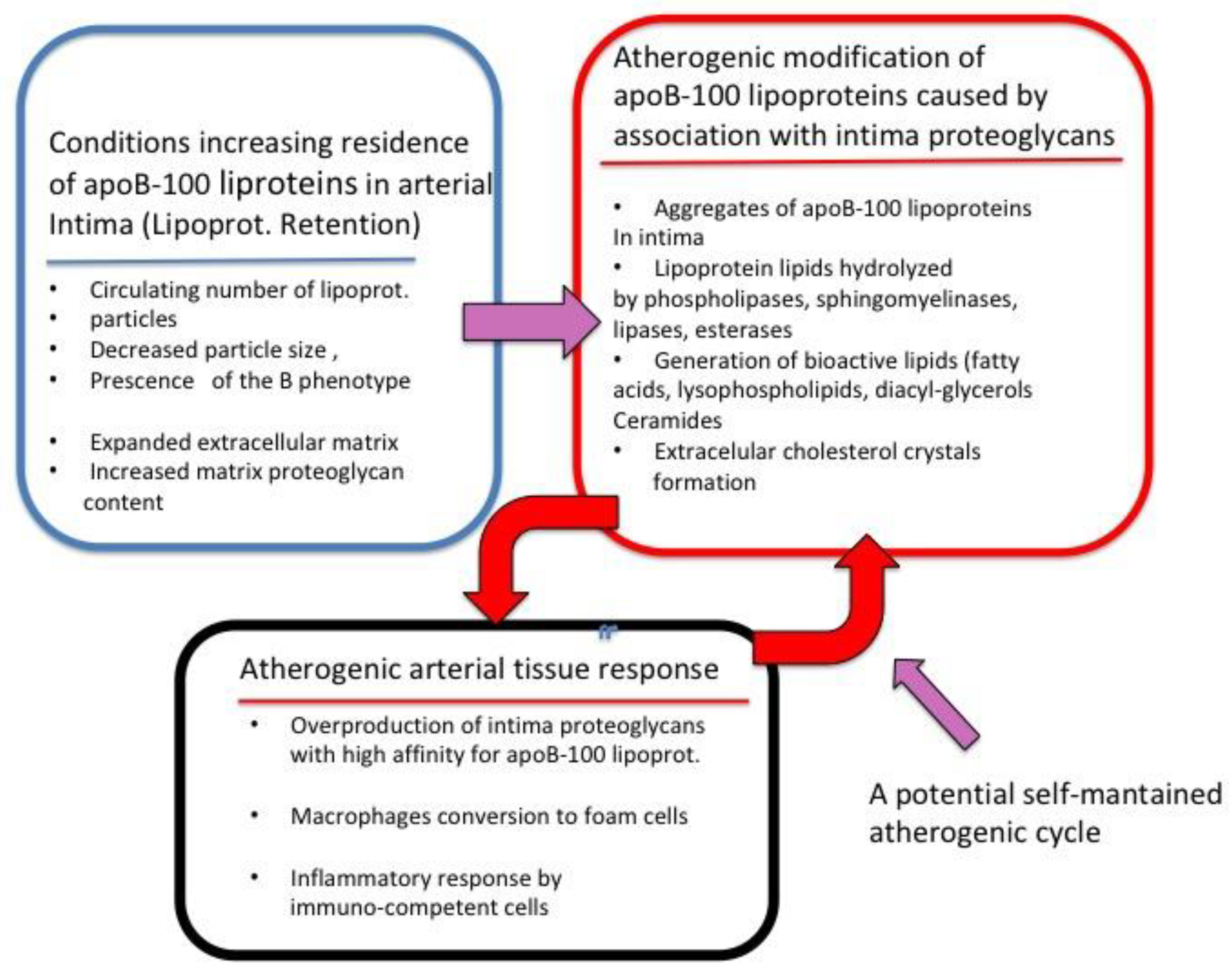
3. ApoB-100 Lipoprotein Entry and Its Proteoglycan-Mediated Retention in the Arterial Intima
4. Atherogenic Structural and Biochemical ApoB-100 Lipoprotein Alterations Caused by PG Retention
5. Cellular Consequences of the Association of ApoB-100 Lipoproteins with Intima Proteoglycans
6. Alterations of the Arterial Intima Extracellular Matrix that May Contribute to Increased Retention of ApoB-100 Lipoproteins
7. Conclusions
Funding
Conflicts of Interest
References
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, H.A.; Krauss, R.A.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Faber, M. The human aorta: Sulfate-containing polyuronides and the deposition of cholesterol. Arch. Pathol. Lab. Med. 1949, 48, 342–350. [Google Scholar] [CrossRef]
- Iverius, P.-H. The interaction between human plasma lipoproteins and commective tissue glycosaminoglycans. J. Biol. Chem. 1972, 247, 2607–2613. [Google Scholar] [PubMed]
- Srinivasan, S.; Dolan, P.; Radhakrishnamurthy, B.; Pargogaonkar, P.S.; Berenson, G.S. Lipoprotein mucopolysaccarides complexes from human atherosclerotic lesion. Biochim. Biophys. Acta 1975, 388, 58–70. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G.; Hurt-Camejo, E.; Wiklund, O.; Bondjers, G. Association of apo B lipoproteins with arterial proteoglycans: Pathological significance and molecular basis. Atherosclerosis 1998, 139, 205–222. [Google Scholar] [CrossRef]
- Borén, J.; Williams, K.J. The Central Role of Arterial retention of Cholesterol-rich apolipoprotein B-containing lipoproteins in the Pathogenesis of Atherosclerosis: A triumph of Simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G.; Hurt-Camejo, E. Macrophages, extracellular matrix, and lipoproteins in arterial cholesterol balance. J. Lipid Res. 2014, 55, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G.; Ponce, E.; Lopez, F.; Starosta, R.; Hurt, E.; Romano, M. Partial structure of the active moiety of a lipoprotein complexing proteoglycan from human aorta. Atherosclerosis 1983, 9, 241–254. [Google Scholar] [CrossRef]
- Bernfeldd, P.; Nisselbaum, J. Reaction of human beta-lipoproteins with macromolecular polysulfated esters. Fed. Proc. 1956, 15, 220–227. [Google Scholar]
- Camejo, G.; Acquatella, H.; LaLaguna, F. The interaction of low density lipoproteins with arterial proteoglycan: An additional risk factor? Atherosclerosis 1980, 36, 55–65. [Google Scholar] [CrossRef]
- Lindén, T.; Bondjers, G.; Camejo, G.; Bergstrand, R.; Wilhensen, L.; Wiklund, O. Affinity of LDL to a human arterial proteoglycan among male survivors of myocardial infarct. Eur. J. Clin. Investig. 1989, 19, 38–44. [Google Scholar] [CrossRef]
- Fagerberg, B.; Wiklund, O.; Agewall, S.; Camejo, G.; Wikstrand, R.J. Multifactorial treatment of hypertensive men at high cardiovascular risk and low-density lipoprotein cholesterol affinity to human arterial proteoglycans. Eur. J. Clin. Investig. 1996, 26, 960–965. [Google Scholar] [CrossRef]
- Wiklund, O.; Bondjers, G.; Wright, I.; Camejo, G. Insoluble complex formation between LDL and arterial proteoglycans in relation to serum lipid levels and effecs of lipid lowering drugs. Atherosclerosis 1996, 119, 57–67. [Google Scholar] [CrossRef]
- Garces, F.; Lopéz, F.; Niño, C.; Fernandéz, A.; Chacin, L.; Hurt-Camejo, E.; Camejo, G.; Apitz, A. High plasma phospholipase A2 activity, inflammation markers, and LDL alterations in obesity with or without type 2 diabetes. Obesity 2010, 18, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteinsin in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Bencells, C.; Benitez, S.; Jauhianien, M.; Ordoñez-Llanos, J.; Kovanen, P.; Villegas, S.; Sanchez-Quesada, L.; Öörni, K. High binding affinity of electronegative LDL to human aortic proteoglycans depends of its aggregation level. J. Lipid Res. 2009, 51, 446–465. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.B.; Slater, R. Relationship between low density lipoprotein in aortic intima and serum lipid levels. Lancet 1972, 299, 463–469. [Google Scholar] [CrossRef]
- Shaikh, M.; Wootton, R.; Nordestgaard, B.G.; Baskerville, P.; Lumley, J.-S.; LaVille, A.E.; Quiney, J.; Lewis, B. Quantitative studies of transfer in vivo of low density, Sf 12-60, and Sf 60-400 lipoproteins between plasma and arterial intima in humans. Arterioscler. Thromb. Vasc. Biol. 1991, 11, 569–577. [Google Scholar] [CrossRef]
- Nakashima, Y.; Fujii, H.; Sumiyoshi, S.; Wight, T.N.; Sueshi, K. Early human atheroclerosis:accumulation of lipids and proteoglycans in intimal thickening followed by macrophage infiltration. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G.; Olofsson, S.; Lopez, F.; Carlsson, P.; Bondjers, G. Identification of Apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycans. Arteriosclerosis 1988, 8, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.; Hulthe, J.; Fagerberg, B.; Olsson, B.-M.; Hallberg, C.; Dahlöf, B.; Camejo, G. A proteomic study of apolipoproteins in LDL subclasses in patients with the metabolic syndrome and type 2 diabetes. J. Lipid Res. 2005, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Olin-Lewis, K.; Krauss, R.M.; La Belle, M.; Blanche, P.J.; Barret, P.H.; Wight, T.N.; Chait, A. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascula proteoglycan biglycan. J. Lipid Res. 2002, 43, 1969–1977. [Google Scholar] [CrossRef]
- Hiukka, A.; Ståhlman, M.; Petterso, C.; Levin, M.; Adiels, M.; Teneberg, S.; Leionen, E.S.; Mattson Hulten, L.; Wiklund, O.; Oresic, M.; et al. ApoCIII-enriched LDL in type 2 diabetes displays altered lipid composition, increased dusceptibility for sphingomyelinase, and increased binding to byglycan. Diabetes 2009, 58, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Li, Y.; Brocia, R.; Xu, S.W.; Swenson, T.L.; Williams, K.J. Lipoprotein lipase and sphingomyelinase synergistically enhance association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoproteins and lipoprotein (a) retention and macrophage foam cell formation. J. Biol. Chem. 1993, 268, 20419–20432. [Google Scholar] [PubMed]
- Sneck, M.; Nguyen, S.D.; Pihiajamas, T.; Yohannes, G.; Riekkola, M.L.; Milne, R.; Kovanen, P.; Öörne, K. Conformational changes of apoB-100 in Smase-modified LL mediate formation of large aggregates at acidic pH. J. Lipid Res. 2012, 53, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Hurt-Camejo, E.; Camejo, G.; Peilot, H.; Öörni, K.; Kovanen, P. Phospholipase A2 in vascular disease. Circ. Res. 2001, 89, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Öörni, K.; Rajamäki, K.; Nguyen, S.D.; Lähdesmäki, K.; Plihtari, R.; Lee-Rueckert, M.; Kovanen, P.T. Acidification of the intimal fluid: The perfect storm for atherosclerosis. J. Lipid Res. 2015, 56, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Hurt-Camejo, E.; Camejo, G.; Rosengren, B.; Lopez, F.; Wiklund, O.; Bondjers, G. Differential uptake of proteoglycan-selected subfractions of low density lipoprotein by human macrophages. J. Lipid Res. 1990, 31, 1387–1398. [Google Scholar] [PubMed]
- Delgado-Roche, I.; Brito, V.; Acosta, E.; Perez, A.; Fernandez, J.R.; Hernandez-Mato, Y.; Griñan, T.; Soto, Y.; León, O.S.; Marleau, S.; et al. Arresting progressive atherosclerosis by immunization with an anti-glycosaminoglycan monoclonal antibody in apolipoprotein E-deficient mice. Free Radic. Biol. Med. 2015, 89, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Mateu, L.; Kirchhausen, T.; Camejo, G. Small-Angle X-Ray scattering and differential scanning calorimetry studies on seversibly modified human-serum low density lipoproteins. Biochemistry 1978, 17, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Camejo, G.; Hurt, E.; Wiklund, O.; Rosengren, B.; Lopez, F.; Bondjers, G. Modifications of low density lipoprotein induced by arterial proteoglycans and chondroitin-6-sulfate. Biochim. Biophys. Acta 1991, 1096, 253–261. [Google Scholar] [CrossRef]
- Nievelstein, P.; Fogelman, A.; Mottino, G. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoproteins: A deep-etch and immunolocalization studyof ultrarapidly frozen tissue. Arter. Thromb. Vasc. Biol. 1991, 1, 1795–1805. [Google Scholar] [CrossRef]
- Hoff, H.; Hoppe, G. Structure of cholesterol-containing particles accumulating in atherosclerotic lesions and the mechanisms of their derivation. Curr. Opin. Lipidol. 1995, 6, 311–325. [Google Scholar] [CrossRef]
- Camejo, G.; Hurt, E.; Romano, M. Properties of lipoprotein complexes isolated by affinity chromatography from human aortas. Biomed. Biochim. Acta 1985, 44, 389–401. [Google Scholar] [PubMed]
- Hurt-camejo, E.; Olsson, U.; Wiklund, O.; Bondjers, G. Cellular consequences of the association of apoB lipoproteins with proteoglycans. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.; Small, D.J.; Li, Z.; Schaefer, E. Differences in LDL subspecies involve alterations in lipid composition and conformational changes in apoB conformation. J. Lipid Res. 1196, 37, 1924–1935. [Google Scholar]
- Berneis, K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Flood, C.; Gustafsson, M.; Pitas, R.E.; Amaboldi, L.; Walzem, R.M.; Borén, J. Molecular mechanisms for changes in proteoglycan binding on compositional changes of core and the surface of Low-density lipoprotein–containing human apolipoprotein apoB100. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Svensson, L.; Hurt-Camejo, E. Phospholipase A2 modifications of low density lipoproteins forms small, high density particles with increased affinity for proteoglycans and glycosaminoglycans. J Biol. Chem. 1999, 274, 25913–25920. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.; Blackenhorn, D.; Chandler, A.; Glagov, S.; Insunll, W.; Richardson, M.; Rosenfeld, M.; Schaffer, S.; Schwarts, C.; Wagner, W.D. A definition of the intima of human arteries and its atherosclerotic-prone regions. Circulation 1992, 85, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Rodriguéz-Lee, M.; Ostergren-Lunden, G.; Wallin, B.; Hurt-Camejo, E.; Bondjers, G.; Camejo, G. Fatty acids cause alterations of human arterial smooth muscle cells that increase the affinity for low density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Rodriguéz-Lee, M.; Bondjer, G.; Camejo, G. Fatty acid induced atherogenic changes in extracellular matrix proteoglycans. Curr. Opin. Lipidol. 2007, 18, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Kijani, S.; Vázquez, A.; Levin, M.; Borén, J.; Fogelstrand, P. Intimal hyperplasia induced by vascular intervention causes lipoprotein retention and accelerated atherosclerosis. Physiol. Rep. 2017, 5, e13334. [Google Scholar] [CrossRef] [PubMed]
- Bach Steffensen, L.; BØdttker Mortensen, M.; Kjolby, M.; Kallestrup Hengensen, M.; Oxvig, C.; Bentzon, J.F. Disturbed laminar blood flow vastly augments lipoprotein retention in the arterial wall: A key mechanism distinguishing susceptible from resistant sites. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1928–1935. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurt-Camejo, E.; Camejo, G. ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker. J. Cardiovasc. Dev. Dis. 2018, 5, 36. https://doi.org/10.3390/jcdd5030036
Hurt-Camejo E, Camejo G. ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker. Journal of Cardiovascular Development and Disease. 2018; 5(3):36. https://doi.org/10.3390/jcdd5030036
Chicago/Turabian StyleHurt-Camejo, Eva, and Germán Camejo. 2018. "ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker" Journal of Cardiovascular Development and Disease 5, no. 3: 36. https://doi.org/10.3390/jcdd5030036
APA StyleHurt-Camejo, E., & Camejo, G. (2018). ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker. Journal of Cardiovascular Development and Disease, 5(3), 36. https://doi.org/10.3390/jcdd5030036