Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System


Abstract
1. Introduction
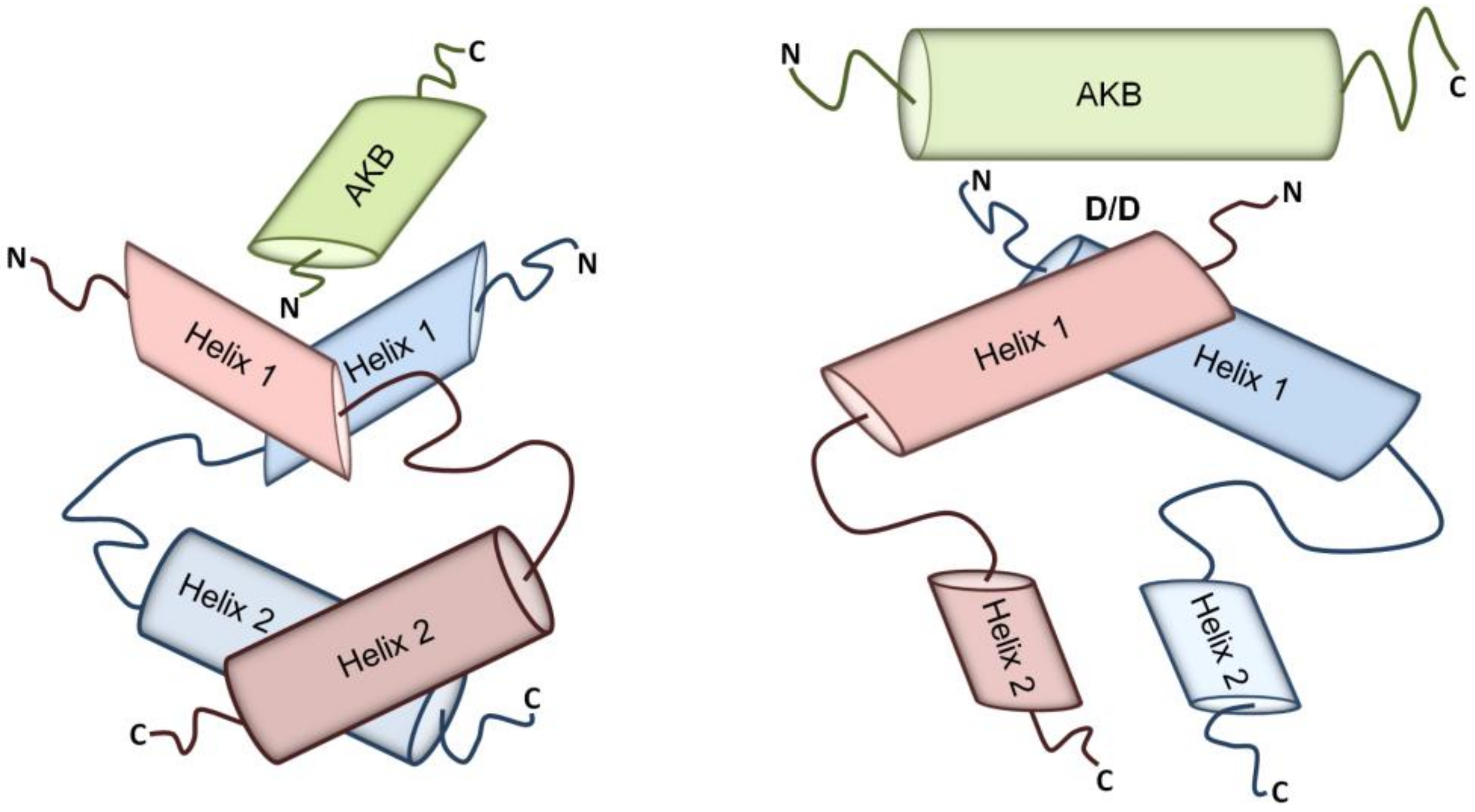
2. A-kinase Anchoring Proteins (AKAPs)
2.1. AKAP Subcellular Localization
2.2. AKAPs in the Cardiovascular System
2.2.1. AKAPs Regulating the Endothelial Barrier Function
2.2.2. AKAPs Regulating the Vascular Tone
2.2.3. AKAPs Controlling Excitation-Contraction Coupling
2.2.4. AKAPs Regulating Cardiac Repolarization
2.2.5. AKAPs Involved in Cardiac Stress Response
2.2.6. AKAPs Involved in the β-ARs Desensitization/Resensitization Cycle
2.3. Aberrant cAMP Compartmentalization Can be Visualized
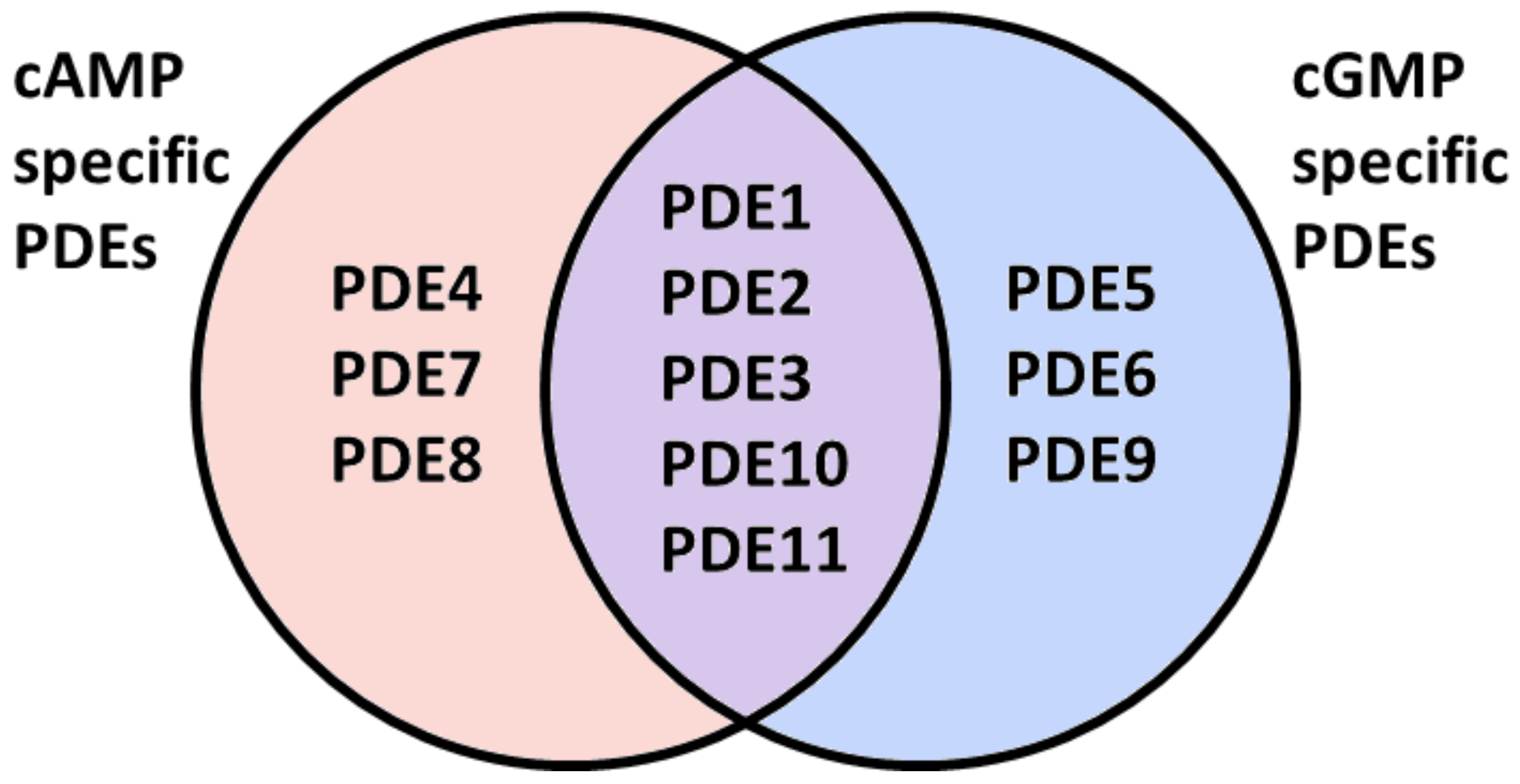
3. Cyclic Nucleotide Phosphodiesterases (PDEs)
3.1. PDE Subcellular Localization
3.2. PDEs in the Cardiovascular System
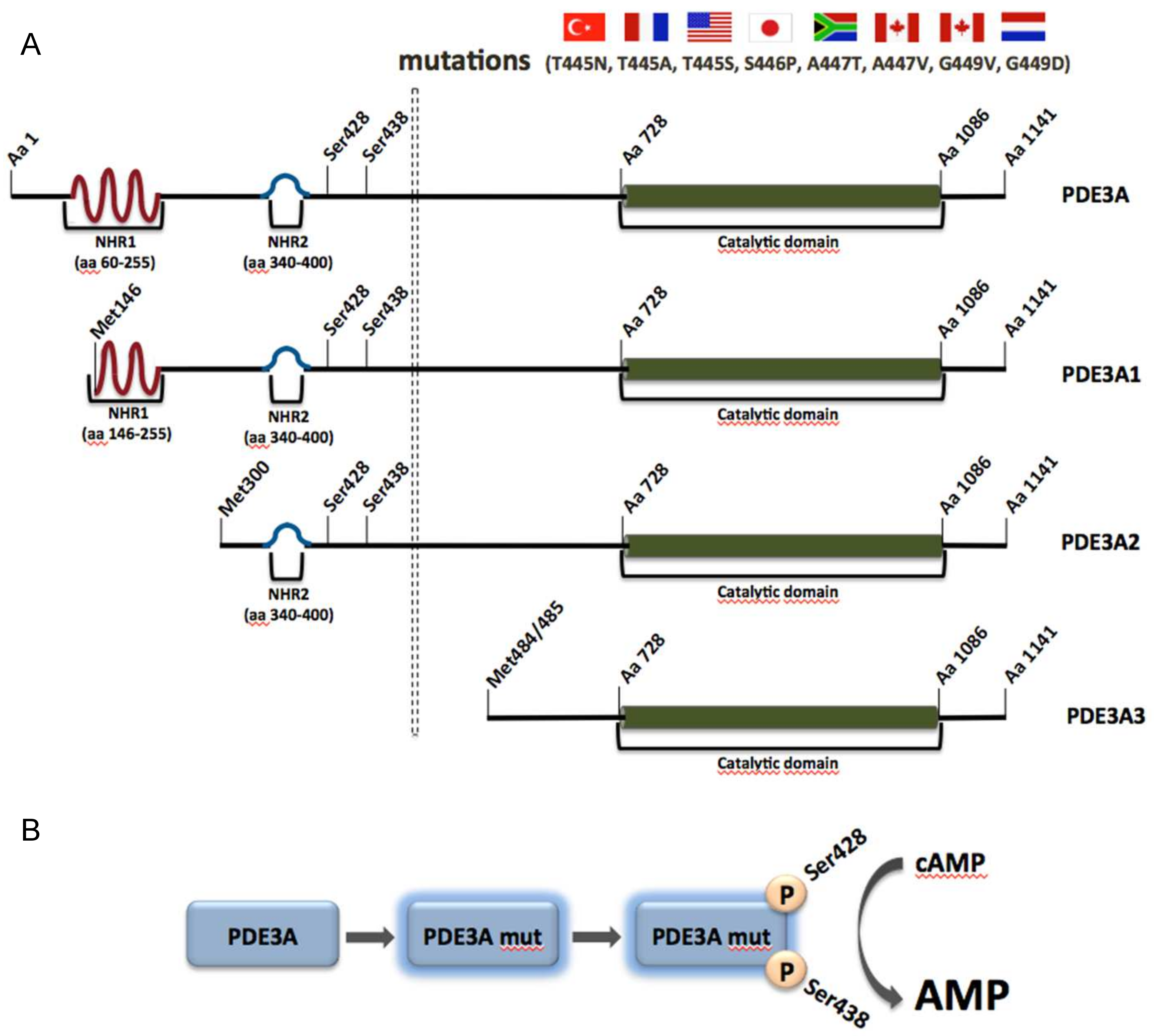
3.2.1. PDE3A and Autosomal Dominant Hypertension with Brachydactyly (HTNB)
3.3. PDE Inhibitors
3.3.1. PDE3
3.3.2. PDE4
3.3.3. PDE5
3.3.4. Potential for PDE1, PDE2, PDE8 and PDE9 Inhibitors
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A.; Hypertension, I.S.o. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Bolívar, J.J. Essential hypertension: An approach to its etiology and neurogenic pathophysiology. Int. J. Hypertens. 2013, 2013, 547809. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Nikolaev, V.O. Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol (Oxf) 2013, 207, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bertinetti, D.; Herberg, F.W. cAMP-dependent protein kinase and cGMP-dependent protein kinase as cyclic nucleotide effectors. Handb. Exp. Pharmacol. 2017, 238, 105–122. [Google Scholar] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP sensor epac proteins and their role in cardiovascular function and disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.; Schindler, R. New kids on the block: The popeye domain containing (popdc) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell Signal. 2017, 40, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Szaszák, M.; Christian, F.; Rosenthal, W.; Klussmann, E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008, 20, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Skroblin, P.; Grossmann, S.; Schafer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of protein kinase A anchoring. Int Rev. Cell Mol. Biol 2010, 283, 235–330. [Google Scholar] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deák, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Pidoux, G.; Taskén, K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 2010, 44, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Taskén, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu Rev. Pharmacol Toxicol 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Zaccolo, M. Microdomains in the Cardiovascular System; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Radeva, M.Y.; Kugelmann, D.; Spindler, V.; Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS One 2014, 9, e106733. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.B.; Choi, Y.K.; Lim, J.J.; Kwon, S.H.; Her, S.; Kim, H.J.; Lim, K.J.; Ahn, J.C.; Kim, Y.M.; Bae, M.K.; et al. AKAP12 regulates vascular integrity in zebrafish. Exp. Mol. Med. 2012, 44, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Johnson, B.D.; Westenbroek, R.E.; Hays, L.G.; Yates, J.R.; Scheuer, T.; Catterall, W.A.; Murphy, B.J. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron 1998, 20, 1017–1026. [Google Scholar] [CrossRef]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Shen, W.; Vandeput, F.; Szabo-Fresnais, N.; Krall, J.; Degerman, E.; Goetz, F.; Klussmann, E.; Movsesian, M.; Manganiello, V. Regulation of sarcoplasmic reticulum Ca2+ atpase 2 (serca2) activity by phosphodiesterase 3a (pde3a) in human myocardium: Phosphorylation-dependent interaction of pde3a1 with serca2. J. Biol. Chem. 2015, 290, 6763–6776. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the kcnq1-kcne1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Katus, H.A.; Olson, E.N.; Hill, J.A. Hypertrophy of the heart: A new therapeutic target? Circulation 2004, 109, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Deák, V.A.; Klussmann, E. Pharmacological interference with protein-protein interactions of akinase anchoring proteins as a strategy for the treatment of disease. Curr. Drug Targets 2016, 17, 1147–1171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.E.; Kass, D.A. Cardiac phosphodiesterases and their modulation for treating heart disease. Handb. Exp. Pharmacol. 2017, 243, 249–269. [Google Scholar] [PubMed]
- Surapisitchat, J.; Jeon, K.I.; Yan, C.; Beavo, J.A. Differential regulation of endothelial cell permeability by cgmp via phosphodiesterases 2 and 3. Circ. Res. 2007, 101, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Špiranec, K.; Röck, K.; Fischer, J.W.; Kämmerer, U.; et al. Endothelial actions of anp enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ. Res. 2016, 119, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Miller, C.L.; Abe, J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ. Res. 2007, 100, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Vargas, M.L.; Kaumann, A.J. Phosphodiesterases PDE3 and PDE4 jointly control the inotropic effects but not chronotropic effects of (-)-cgp12177 despite PDE4-evoked sinoatrial bradycardia in rat atrium. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 379, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.I.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Oikawa, M.; Cai, Y.; Wojtovich, A.P.; Nagel, D.J.; Xu, X.; Xu, H.; Florio, V.; Rybalkin, S.D.; Beavo, J.A.; et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 2009, 105, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Belacel-Ouari, M.; Bedioune, I.; Zhang, L.; Leroy, J.; Leblais, V.; Fischmeister, R.; Vandecasteele, G. Cyclic nucleotide phosphodiesterases in heart and vessels: A therapeutic perspective. Arch. Cardiovasc. Dis. 2016, 109, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Singh, S.; Suryavanshi, S.V.; Altarabsheh, S.E.; Deo, S.V.; McConnell, B.K. Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 2014, 16, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.J.; Jones, B.W.; Scott, J.D. Networking with AKAPs: Context-dependent regulation of anchored enzymes. Mol. Interv. 2010, 10, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nucleotide-dependent protein kinases. Annu. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Ilouz, R.; Zhang, P.; Kornev, A.P. Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Walker-Gray, R.; Stengel, F.; Gold, M.G. Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc. Natl. Acad. Sci. USA 2017, 114, 10414–10419. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Fletcher, W.H.; Johnson, D.A. Regulation of cAMP-dependent protein kinase: Enzyme activation without dissociation. Biochemistry 1995, 34, 6267–6271. [Google Scholar] [CrossRef] [PubMed]
- Kopperud, R.; Christensen, A.E.; Kjarland, E.; Viste, K.; Kleivdal, H.; Doskeland, S.O. Formation of inactive cAMP-saturated holoenzyme of cAMP-dependent protein kinase under physiological conditions. J. Biol. Chem. 2002, 277, 13443–13448. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Ruehr, M.L.; Zakhary, D.R.; Damron, D.S.; Bond, M. Cyclic amp-dependent protein kinase binding to A-kinase anchoring proteins in living cells by fluorescence resonance energy transfer of green fluorescent protein fusion proteins. J. Biol. Chem. 1999, 274, 33092–33096. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Tasken, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular basis of akap specificity for PKA regulatory subunits. Mol. Cell 2006, 24, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.H.; Jennings, P.A.; Taylor, S.S. A dynamic mechanism for akap binding to rii isoforms of cAMP-dependent protein kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Götz, F.; Roske, Y.; Schulz, M.S.; Autenrieth, K.; Bertinetti, D.; Faelber, K.; Zühlke, K.; Kreuchwig, A.; Kennedy, E.J.; Krause, G.; et al. AKAP18:Pka-riiα structure reveals crucial anchor points for recognition of regulatory subunits of PKA. Biochem. J. 2016, 473, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- McSorley, T.; Stefan, E.; Henn, V.; Wiesner, B.; Baillie, G.S.; Houslay, M.D.; Rosenthal, W.; Klussmann, E. Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur. J. Cell. Biol. 2006, 85, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. Identification of a novel protein kinase A anchoring protein that binds both type i and type ii regulatory subunits. J. Biol. Chem. 1997, 272, 8057–8064. [Google Scholar] [CrossRef] [PubMed]
- Kovanich, D.; van der Heyden, M.A.; Aye, T.T.; van Veen, T.A.; Heck, A.J.; Scholten, A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type i cAMP-dependent protein kinase. Chembiochem 2010, 11, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type i a-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.P.; Ma, Y.; Margarucci, L.; Mackey, M.; van der Heyden, M.A.; Ellisman, M.; Scholten, A.; Taylor, S.S.; Heck, A.J. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit i (PKA-ri) to the plasma membrane. J. Biol. Chem. 2012, 287, 43789–43797. [Google Scholar] [CrossRef] [PubMed]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Fraser, I.D.; Tavalin, S.J.; Lester, L.B.; Langeberg, L.K.; Westphal, A.M.; Dean, R.A.; Marrion, N.V.; Scott, J.D. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998, 17, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Trotter, K.W.; Fraser, I.D.; Scott, G.K.; Stutts, M.J.; Scott, J.D.; Milgram, S.L. Alternative splicing regulates the subcellular localization of A-kinase anchoring protein 18 isoforms. J. Cell Biol. 1999, 147, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Dell’Acqua, M.L.; Faux, M.C.; Thorburn, J.; Thorburn, A.; Scott, J.D. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 1998, 17, 2246–2260. [Google Scholar] [CrossRef] [PubMed]
- Hundsrucker, C.; Skroblin, P.; Christian, F.; Zenn, H.M.; Popara, V.; Joshi, M.; Eichhorst, J.; Wiesner, B.; Herberg, F.W.; Reif, B.; et al. Glycogen synthase kinase 3β interaction protein functions as an A-kinase anchoring protein. J. Biol. Chem. 2010, 285, 5507–5521. [Google Scholar] [CrossRef] [PubMed]
- Deák, V.A.; Skroblin, P.; Dittmayer, C.; Knobeloch, K.P.; Bachmann, S.; Klussmann, E. The A-kinase anchoring protein gskip regulates gsk3β activity and controls palatal shelf fusion in mice. J. Biol. Chem. 2016, 291, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Schröter, M.F.; Perets, E.; Skroblin, P.; Moutty, M.C.; Deàk, V.A.; Birchmeier, W.; Klussmann, E. The A-kinase anchoring protein (AKAP) glycogen synthase kinase 3β interaction protein (gskip) regulates β-catenin through its interactions with both protein kinase a (PKA) and gsk3β. J. Biol. Chem. 2016, 291, 19618–19630. [Google Scholar] [CrossRef] [PubMed]
- Scholten, A.; Poh, M.K.; van Veen, T.A.; van Breukelen, B.; Vos, M.A.; Heck, A.J. Analysis of the cGMP/cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J. Proteom. Res. 2006, 5, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase a. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Wang, L.; Ma, Y.; Durick, K.; Perkins, G.; Deerinck, T.J.; Ellisman, M.H.; Taylor, S.S. Nh2-terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (d-AKAP1) to either mitochondria or endoplasmic reticulum. J. Cell. Biol 1999, 145, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Langeberg, L.K.; Doxsey, S.J.; Scott, J.D. Pericentrin anchors protein kinase a at the centrosome through a newly identified rii-binding domain. Curr. Biol. 2000, 10, 417–420. [Google Scholar] [CrossRef]
- Gillingham, A.K.; Munro, S. The pact domain, a conserved centrosomal targeting motif in the coiled-coil proteins AKAP450 and pericentrin. EMBO Rep. 2000, 1, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Santana, L.F. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Santana, L.F. Cav1.2 sparklets in heart and vascular smooth muscle. J. Mol. Cell. Cardiol. 2013, 58, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Mercado, J.; Baylie, R.; Navedo, M.F.; Yuan, C.; Scott, J.D.; Nelson, M.T.; Brayden, J.E.; Santana, L.F. Local control of trpv4 channels by AKAP150-targeted PKC in arterial smooth muscle. J. Gen. Physiol. 2014, 143, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Hulme, J.T.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac cav1.2 channels during β1-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 16574–16579. [Google Scholar] [CrossRef] [PubMed]
- Appert-Collin, A.; Cotecchia, S.; Nenniger-Tosato, M.; Pedrazzini, T.; Diviani, D. The A-kinase anchoring protein (AKAP)-lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl Acad Sci USA 2007, 104, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase a anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Shumay, E.; Wang, H.; Malbon, C.C. The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the β2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J. Biol. Chem. 2001, 276, 24005–24014. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.A.; Tavalin, S.J.; Goehring, A.S.; Scott, J.D.; Bahouth, S.W. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the β1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J. Biol. Chem. 2006, 281, 33537–33553. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Cinel, I.; Dellinger, R.P. Advances in pathogenesis and management of sepsis. Curr. Opin. Infect. Dis. 2007, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, J.H.; Kim, W.J.; Lee, H.Y.; Park, J.A.; Lee, S.W.; Yoon, D.K.; Kim, H.H.; Chung, H.; Yu, Y.S.; et al. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1α. J. Neurosci. 2007, 27, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, N.; Waschke, J. cAMP with other signaling cues converges on rac1 to stabilize the endothelial barrier—A signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, S.; Ernandez, T.; Cullere, X.; Takahashi, M.; Ono, Y.; Komarova, Y.; Mayadas, T.N. AKAP9 regulation of microtubule dynamics promotes epac1-induced endothelial barrier properties. Blood 2011, 117, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Nieves-Cintron, M.; Amberg, G.C.; Yuan, C.; Votaw, V.S.; Lederer, W.J.; McKnight, G.S.; Santana, L.F. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin ii-induced hypertension. Circ. Res. 2008, 102, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Cheng, E.P.; Yuan, C.; Votaw, S.; Molkentin, J.D.; Scott, J.D.; Santana, L.F. Increased coupled gating of l-type Ca2+ channels during hypertension and timothy syndrome. Circ. Res. 2010, 106, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.E.; Cheng, E.P.; Mercado, J.L.; Santana, L.F. L-type Ca2+ channel function during timothy syndrome. Trends Cardiovasc. Med. 2012, 22, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Heppner, T.J.; Nelson, M.T.; Brayden, J.E. Trpv4 forms a novel Ca2+ signaling complex with ryanodine receptors and bkca channels. Circ. Res. 2005, 97, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Pauyo, T.; Drapp, R.; Tavares, M.J.; Liedtke, W.; Brayden, J.E. Trpv4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1096–H1102. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Szentesi, P.; Pignier, C.; Egger, M.; Kranias, E.G.; Niggli, E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+-induced Ca2+ release refractoriness in heart muscle. Circ. Res. 2004, 95, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Kranias, E.G.; Hajjar, R.J. Modulation of cardiac contractility by the phospholamban/serca2a regulatome. Circ. Res. 2012, 110, 1646–1660. [Google Scholar] [CrossRef] [PubMed]
- Bünemann, M.; Gerhardstein, B.L.; Gao, T.; Hosey, M.M. Functional regulation of l-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J. Biol. Chem. 1999, 274, 33851–33854. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.B.; Rossow, C.F.; Navedo, M.F.; Westenbroek, R.E.; Catterall, W.A.; Santana, L.F.; McKnight, G.S. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of l-type calcium channels. Circ. Res. 2010, 107, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Schulze, D.H.; Muqhal, M.; Lederer, W.J.; Ruknudin, A.M. Sodium/calcium exchanger (ncx1) macromolecular complex. J. Biol. Chem. 2003, 278, 28849–28855. [Google Scholar] [CrossRef] [PubMed]
- Ruknudin, A.; He, S.; Lederer, W.J.; Schulze, D.H. Functional differences between cardiac and renal isoforms of the rat Na+-Ca2+ exchanger ncx1 expressed in xenopus oocytes. J. Physiol. 2000, 529, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Nicodemus-Johnson, J.; Carnegie, G.K.; Danziger, R.S. Molecular evolution of A-kinase anchoring protein (AKAP)-7: Implications in comparative PKA compartmentalization. BMC Evol. Biol. 2012, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Kass, R.S. Recent progress in congenital long qt syndrome. Curr. Opin. Cardiol. 2010, 25, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S.; Burns-Hamuro, L.L.; Ma, Y.; Hamon, S.C.; Canaves, J.M.; Shi, M.M.; Nelson, M.R.; Sing, C.F.; Cantor, C.R.; Taylor, S.S.; et al. Amino acid variant in the kinase binding domain of dual-specific a kinase-anchoring protein 2: A disease susceptibility polymorphism. Proc. Natl. Acad. Sci. USA 2003, 100, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Tingley, W.G.; Pawlikowska, L.; Zaroff, J.G.; Kim, T.; Nguyen, T.; Young, S.G.; Vranizan, K.; Kwok, P.Y.; Whooley, M.A.; Conklin, B.R. Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Łoniewska, B.; Kaczmarczyk, M.; Clark, J.S.; Gorący, I.; Horodnicka-Józwa, A.; Ciechanowicz, A. Association of functional genetic variants of A-kinase anchoring protein 10 with qt interval length in full-term polish newborns. Arch. Med. Sci. 2015, 11, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-lbc anchors protein kinase A and nucleates Gα12-selective rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E.; Edemir, B.; Pepperle, B.; Tamma, G.; Henn, V.; Klauschenz, E.; Hundsrucker, C.; Maric, K.; Rosenthal, W. Ht31: The first protein kinase A anchoring protein to integrate protein kinase A and rho signaling. FEBS Lett. 2001, 507, 264–268. [Google Scholar] [CrossRef]
- Abdul Azeez, K.R.; Knapp, S.; Fernandes, J.M.; Klussmann, E.; Elkins, J.M. The crystal structure of the rhoa-AKAP-lbc dh-ph domain complex. Biochem. J. 2014, 464, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Schrade, K.; Tröger, J.; Eldahshan, A.; Zühlke, K.; Abdul Azeez, K.R.; Elkins, J.M.; Neuenschwander, M.; Oder, A.; Elkewedi, M.; Jaksch, S.; et al. An AKAP-lbc-rhoa interaction inhibitor promotes the translocation of aquaporin-2 to the plasma membrane of renal collecting duct principal cells. PLoS One 2018, 13, e0191423. [Google Scholar] [CrossRef] [PubMed]
- Mayers, C.M.; Wadell, J.; McLean, K.; Venere, M.; Malik, M.; Shibata, T.; Driggers, P.H.; Kino, T.; Guo, X.C.; Koide, H.; et al. The rho guanine nucleotide exchange factor AKAP13 (brx) is essential for cardiac development in mice. J. Biol. Chem. 2010, 285, 12344–12354. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Abuin, L.; Cotecchia, S.; Pansier, L. Anchoring of both PKA and 14-3-3 inhibits the rho-gef activity of the AKAP-lbc signaling complex. EMBO J. 2004, 23, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Smith, F.D.; McConnachie, G.; Langeberg, L.K.; Scott, J.D. AKAP-lbc nucleates a protein kinase D activation scaffold. Mol. Cell 2004, 15, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 2008, 32, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pare, G.C.; Easlick, J.L.; Mislow, J.M.; McNally, E.M.; Kapiloff, M.S. Nesprin-1alpha contributes to the targeting of mAKAP to the cardiac myocyte nuclear envelope. Exp. Cell Res. 2005, 303, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Kelley, G.G.; Kapiloff, M.S.; Smrcka, A.V. Phospholipase c epsilon scaffolds to muscle-specific a kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J. Biol. Chem. 2011, 286, 23012–23021. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Bauman, A.; Mayer, N.; Henson, E.; Heredia, L.; Ahn, J.; McAvoy, T.; Nairn, A.C.; Kapiloff, M.S. Camp-stimulated protein phosphatase 2a activity associated with muscle a kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase pde4d3. J. Biol. Chem. 2010, 285, 11078–11086. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Goehring, A.S.; Kapiloff, M.S.; Langeberg, L.K.; Scott, J.D. MAKAP compartmentalizes oxygen-dependent control of hif-1α. Sci. Signal. 2008, 1, ra18. [Google Scholar] [CrossRef] [PubMed]
- Abrenica, B.; AlShaaban, M.; Czubryt, M.P. The a-kinase anchor protein AKAP121 is a negative regulator of cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol 2009, 46, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Xiao, C.Y.; Li, Z.; Zhang, T.; Omens, J.H.; Ishii, I.; Chun, J.; Brown, J.H. Sphingosine 1-phosphate s1p2 and s1p3 receptor-mediated akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2944–H2951. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. AKAP1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS One 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed]
- Lacaná, E.; Maceyka, M.; Milstien, S.; Spiegel, S. Cloning and characterization of a protein kinase a anchoring protein (AKAP)-related protein that interacts with and regulates sphingosine kinase 1 activity. J. Biol. Chem. 2002, 277, 32947–32953. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, N.T.; Mohan, M.L.; Goswami, S.K.; Naga Prasad, S.V. Regulation of β-adrenergic receptor function: An emphasis on receptor resensitization. Cell Cycle 2011, 10, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of β-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef] [PubMed]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Nikolaev, V.O. Imaging alterations of cardiomyocyte cAMP microdomains in disease. Front. Pharmacol. 2015, 6, 172. [Google Scholar] [CrossRef] [PubMed]
- Pavlaki, N.; Nikolaev, V.O. Imaging of PDE2- and PDE3-mediated cGMP-to-cAMP cross-talk in cardiomyocytes. J. Cardiovasc. Dev. Dis. 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Musheshe, N.; Schmidt, M.; Zaccolo, M. cAMP: From long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 2017, 39, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Thestrup, T.; Litzlbauer, J.; Bartholomäus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and t lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical techniques for detection of cAMP and cGMP in living cells. Int J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.K.; Sprenger, J.U.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to anp-induced augmentation of β-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Jungen, C.; Scherschel, K.; Eickholt, C.; Kuklik, P.; Klatt, N.; Bork, N.; Salzbrunn, T.; Alken, F.; Angendohr, S.; Klene, C.; et al. Disruption of cardiac cholinergic neurons enhances susceptibility to ventricular arrhythmias. Nat. Commun. 2017, 8, 14155. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, J.L.; Bhargava, A.; O’Hara, T.; Glukhov, A.V.; Schobesberger, S.; Bhogal, N.; Sikkel, M.B.; Mansfield, C.; Korchev, Y.E.; Lyon, A.R.; et al. Microdomain-specific modulation of l-type calcium channels leads to triggered ventricular arrhythmia in heart failure. Circ. Res. 2016, 119, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Rieken, F.; Wagner, J.; Sungkaworn, T.; Zabel, U.; Borzi, A.; Cocucci, E.; Zürn, A.; Lohse, M.J. Single-molecule analysis of fluorescently labeled g-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl Acad Sci USA 2013, 110, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Miragoli, M.; Moshkov, A.; Novak, P.; Shevchuk, A.; Nikolaev, V.O.; El-Hamamsy, I.; Potter, C.M.; Wright, P.; Kadir, S.H.; Lyon, A.R.; et al. Scanning ion conductance microscopy: A convergent high-resolution technology for multi-parametric analysis of living cardiovascular cells. J. R Soc. Interface 2011, 8, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Palmer, D.; Tilley, D.G.; Dunkerley, H.A.; Netherton, S.J.; Raymond, D.R.; Elbatarny, H.S.; Jimmo, S.L. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol. Pharmacol. 2003, 64, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Anant, J.S.; Ong, O.C.; Xie, H.Y.; Clarke, S.; O’Brien, P.J.; Fung, B.K. In vivo differential prenylation of retinal cyclic gmp phosphodiesterase catalytic subunits. J. Biol. Chem. 1992, 267, 687–690. [Google Scholar] [PubMed]
- Baillie, G.S.; MacKenzie, S.J.; McPhee, I.; Houslay, M.D. Sub-family selective actions in the ability of erk2 map kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br. J. Pharmacol. 2000, 131, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of pdes and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Wang, H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr. Top. Med. Chem. 2007, 7, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.; Choi, Y.H.; Krall, J.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase PDE3a in cardiac myocytes. J. Biol. Chem. 2002, 277, 38072–38078. [Google Scholar] [CrossRef] [PubMed]
- Senzaki, H.; Smith, C.J.; Juang, G.J.; Isoda, T.; Mayer, S.P.; Ohler, A.; Paolocci, N.; Tomaselli, G.F.; Hare, J.M.; Kass, D.A. Cardiac phosphodiesterase 5 (cGMP-specific) modulates β-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001, 15, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cdnas encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.H.; Zhang, K.; Chen, C.K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cgmp phosphodiesterase delta subunit (pdedelta) functions as a prenyl-binding protein. J. Biol. Chem. 2004, 279, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Sonati, P.; Rubin, C.; Michaeli, T. Pde7a1, a cAMP-specific phosphodiesterase, inhibits cAMP-dependent protein kinase by a direct interaction with c. J. Biol. Chem. 2006, 281, 15050–15057. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, P.; Egan, R.W.; Billah, M.M. Identification and characterization of a new human type 9 cGMP-specific phosphodiesterase splice variant (PDE9a5). Differential tissue distribution and subcellular localization of PDE9a variants. Gene 2003, 314, 15–27. [Google Scholar] [CrossRef]
- Sayner, S.; Stevens, T. Soluble adenylate cyclase reveals the significance of compartmentalized cAMP on endothelial cell barrier function. Biochem. Soc. Trans. 2006, 34, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Creighton, J.; Zhu, B.; Alexeyev, M.; Stevens, T. Spectrin-anchored phosphodiesterase 4d4 restricts cAMP from disrupting microtubules and inducing endothelial cell gap formation. J. Cell. Sci. 2008, 121, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an no/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4d regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of l-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Melenovsky, V.; Marhin, T.; Fitzgerald, P.; Kass, D.A. Sildenafil inhibits β-adrenergic-stimulated cardiac contractility in humans. Circulation 2005, 112, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8a (PDE8a) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Tovar, A.; Kaumann, A.J. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (−)-adrenaline mediated through mouse cardiac β(1)-adrenoceptors. Br. J. Pharmacol. 2008, 153, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (icer) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4b in the cardiac l-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4d deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pokreisz, P.; Vandenwijngaert, S.; Bito, V.; Van den Bergh, A.; Lenaerts, I.; Busch, C.; Marsboom, G.; Gheysens, O.; Vermeersch, P.; Biesmans, L.; et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009, 119, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9a controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, R.; Krall, J.; Tikishvili, E.; Honeggar, M.; Ahmad, F.; Manganiello, V.C.; Movsesian, M.A. Isoforms of cyclic nucleotide phosphodiesterase pde3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J. Biol. Chem. 2005, 280, 39168–39174. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schächterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3a mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hattenbach, L.O.; Toka, H.R.; Toka, O.; Schuster, H.; Luft, F.C. Absence of hypertensive retinopathy in a turkish kindred with autosomal dominant hypertension and brachydactyly. Br. J. Ophthalmol. 1998, 82, 1363–1365. [Google Scholar] [CrossRef] [PubMed]
- Toka, O.; Tank, J.; Schächterle, C.; Aydin, A.; Maass, P.G.; Elitok, S.; Bartels-Klein, E.; Hollfinger, I.; Lindschau, C.; Mai, K.; et al. Clinical effects of phosphodiesterase 3a mutations in inherited hypertension with brachydactyly. Hypertension 2015, 66, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M. Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol. Ther. 2016, 163, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M.; Ahmad, F.; Hirsch, E. Functions of PDE3 isoforms in cardiac muscle. J. Cardiovasc. Dev. Dis 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Conti, M.; Houslay, M.D. Handbook of Experimental Pharmacology 204. In Phosphodiesterases as Drug Targets; Springer International Publishing: Cham, Switzerland, 2011. [Google Scholar]
- Knight, W.; Yan, C. Therapeutic potential of pde modulation in treating heart disease. Future Med. Chem. 2013, 5, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shakur, Y.; Kambayashi, J. Phosphodiesterases as targets for intermittent claudication. Handb Exp. Pharmacol. 2011, 211–236. [Google Scholar]
- Rogers, K.C.; Oliphant, C.S.; Finks, S.W. Clinical efficacy and safety of cilostazol: A critical review of the literature. Drugs 2015, 75, 377–395. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, W.R.; Money, S.R.; Brass, E.P. Long-term safety of cilostazol in patients with peripheral artery disease: The castle study (cilostazol: A study in long-term effects). J. Vasc. Surg. 2008, 47, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Takigawa, T.; Matsumaru, Y.; Hayakawa, M.; Nemoto, S.; Matsumura, A. Cilostazol reduces restenosis after carotid artery stenting. J. Vasc. Surg. 2010, 51, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Cone, J.; Wang, S.; Tandon, N.; Fong, M.; Sun, B.; Sakurai, K.; Yoshitake, M.; Kambayashi, J.; Liu, Y. Comparison of the effects of cilostazol and milrinone on intracellular cAMP levels and cellular function in platelets and cardiac cells. J. Cardiovasc. Pharmacol. 1999, 34, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Faxon, D.P.; Creager, M.A.; Smith, S.C.; Pasternak, R.C.; Olin, J.W.; Bettmann, M.A.; Criqui, M.H.; Milani, R.V.; Loscalzo, J.; Kaufman, J.A.; et al. Atherosclerotic vascular disease conference: Executive summary: Atherosclerotic vascular disease conference proceeding for healthcare professionals from a special writing group of the american heart association. Circulation 2004, 109, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Momi, S.; Falcinelli, E. Anti-platelet therapy: Phosphodiesterase inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Sahin, M.; Alizade, E.; Pala, S.; Alici, G.; Ozkan, B.; Akgun, T.; Emiroglu, Y.; Demir, S.; Yazicioglu, M.V.; Turkmen, M.M. The effect of cilostazol on right heart function and pulmonary pressure. Cardiovasc. Ther. 2013, 31, e88–e93. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Toga, K.; Sudo, T.; Tachibana, K.; Tochizawa, S.; Kimura, Y.; Yoshida, Y.; Hidaka, H. Suppression of arterial intimal hyperplasia by cilostamide, a cyclic nucleotide phosphodiesterase 3 inhibitor, in a rat balloon double-injury model. Br. J. Pharmacol. 2000, 130, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.B.; Dzavík, V. Inotropes and vasopressors: Review of physiology and clinical use in cardiovascular disease. Circulation 2008, 118, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Tariq, S.; Aronow, W.S. Use of inotropic agents in treatment of systolic heart failure. Int J. Mol. Sci. 2015, 16, 29060–29068. [Google Scholar] [CrossRef] [PubMed]
- Movsesian, M. New pharmacologic interventions to increase cardiac contractility: Challenges and opportunities. Curr. Opin. Cardiol. 2015, 30, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, E. Protein-protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell Signal. 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Sims, C.R.; Singh, S.P.; Mu, S.; Gokden, N.; Zakaria, D.; Nguyen, T.C.; Mayeux, P.R. Rolipram improves outcome in a rat model of infant sepsis-induced cardiorenal syndrome. Front. Pharmacol. 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Samardzic, H.; Adamson, R.H.; Renkin, E.M.; Clark, J.F.; Reed, R.K.; Curry, F.R. Phosphodiesterase 4 inhibition attenuates atrial natriuretic peptide-induced vascular hyperpermeability and loss of plasma volume. J. Physiol. 2011, 589, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br. J. Pharmacol. 2011, 163, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Tenor, H.; Hatzelmann, A.; Beume, R.; Lahu, G.; Zech, K.; Bethke, T.D. Pharmacology, clinical efficacy, and tolerability of phosphodiesterase-4 inhibitors: Impact of human pharmacokinetics. Handb Exp. Pharmacol. 2011, 85–119. [Google Scholar]
- Sakkas, L.I.; Mavropoulos, A.; Bogdanos, D.P. Phosphodiesterase 4 inhibitors in immune-mediated diseases: Mode of action, clinical applications, current and future perspectives. Curr. Med. Chem. 2017, 24, 3054–3067. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Apremilast: A review in psoriasis and psoriatic arthritis. Drugs 2017, 77, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B.; et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Yan, M.; Chen, J.; Chaugai, S.; Chen, C.; Wang, D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor β-induced smad signaling. Front. Med. 2014, 8, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; van Heerebeek, L.; Paulus, W.J. Phosphodiesterase-5 inhibition in heart failure with preserved ejection fraction: Trading therapy for prevention. Eur J. Heart Fail. 2017, 19, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Balidemaj, K.; Homma, S.; Wu, H.; Wang, J.; Maybaum, S. Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow-mediated vasodilation in patients with chronic heart failure. J. Am. Coll. Cardiol. 2000, 36, 845–851. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.N.; Das, A.; Koka, S.; Ockaili, R.A.; Xi, L. Emerging new uses of phosphodiesterase-5 inhibitors in cardiovascular diseases. Exp. Clin. Cardiol. 2011, 16, e30–35. [Google Scholar] [PubMed]
- Pofi, R.; Gianfrilli, D.; Badagliacca, R.; Di Dato, C.; Venneri, M.A.; Giannetta, E. Everything you ever wanted to know about phosphodiesterase 5 inhibitors and the heart (but never dared ask): How do they work? J. Endocrinol. Invest. 2016, 39, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, W.K.; Seger, D.; Beavo, J.A. Molecular cloning of a cdna encoding the “61-kda” Calmodulin-stimulated cyclic nucleotide phosphodiesterase. Tissue-specific expression of structurally related isoforms. J. Biol. Chem. 1993, 268, 645–652. [Google Scholar] [PubMed]
- Goraya, T.A.; Cooper, D.M. Ca2+-calmodulin-dependent phosphodiesterase (pde1): Current perspectives. Cell Signal. 2005, 17, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Adachi, H.; Takase, Y.; Yoshitake, S.; Souda, S.; Saito, I. A selective type v phosphodiesterase inhibitor, e4021, dilates porcine large coronary artery. J. Pharmacol. Exp. Ther. 1995, 272, 825–831. [Google Scholar] [PubMed]
- Nagel, D.J.; Aizawa, T.; Jeon, K.I.; Liu, W.; Mohan, A.; Wei, H.; Miano, J.M.; Florio, V.A.; Gao, P.; Korshunov, V.A.; et al. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1a in vascular smooth muscle cell growth and survival. Circ. Res. 2006, 98, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Pullamsetti, S.S.; Kwapiszewska, G.; Dumitrascu, R.; Tian, X.; Weissmann, N.; Ghofrani, H.A.; Kaulen, C.; Dunkern, T.; Schudt, C.; et al. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: Target for reverse-remodeling therapy. Circulation 2007, 115, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Fischmeister, R.; Castro, L.; Abi-Gerges, A.; Rochais, F.; Vandecasteele, G. Species- and tissue-dependent effects of no and cyclic gmp on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 142, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lämmle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S.; et al. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Rentero, C.; Monfort, A.; Puigdomènech, P. Identification and distribution of different mrna variants produced by differential splicing in the human phosphodiesterase 9a gene. Biochem. Biophys. Res. Commun. 2003, 301, 686–692. [Google Scholar] [CrossRef]
- Christian, F.; Szaszák, M.; Friedl, S.; Drewianka, S.; Lorenz, D.; Goncalves, A.; Furkert, J.; Vargas, C.; Schmieder, P.; Götz, F.; et al. Small molecule AKAP-protein kinase a (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J. Biol. Chem. 2011, 286, 9079–9096. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Munir, M.; Aiman, S.; Wadood, A.; Khan, A.U. The in silico identification of small molecules for protein-protein interaction inhibition in AKAP-lbc-rhoa signaling complex. Comput. Biol. Chem. 2017, 67, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Raimondi, F.; Del Vescovo, C.D.; Dreyer, E.; Reggi, E.; Osman, H.; Ruggieri, L.; Gonano, C.; Cavin, S.; Box, C.L.; et al. Small-molecule protein-protein interaction inhibitor of oncogenic rho signaling. Cell Chem. Biol. 2016, 23, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Serrels, B.; Sandilands, E.; Serrels, A.; Baillie, G.; Houslay, M.D.; Brunton, V.G.; Canel, M.; Machesky, L.M.; Anderson, K.I.; Frame, M.C. A complex between fak, rack1, and PDE4d5 controls spreading initiation and cancer cell polarity. Curr. Biol. 2010, 20, 1086–1092. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Common Name | Gene Name | Alternative Name | Regulated Cardiovascular Process |
|---|---|---|---|
| D-AKAP1 | AKAP1 | AKAP121/ AKAP149/AKAP84 | Cardiac stress response |
| D-AKAP2 | AKAP10 | - | Cardiac repolarization |
| AKAP9 (long isoform) | AKAP9 | - | Endothelial barrier function |
| AKAP18α AKAP18γ AKAP18δ | AKAP7 | - | Excitation-contraction coupling |
| AKAP79 | AKAP5 | AKAP75/AKAP150 | Vascular tone; Excitation-contraction coupling; β-AR desensitization/resensitization cycle |
| AKAP220 | AKAP11 | - | Endothelial barrier function |
| AKAP-Lbc | AKAP13 | Brx-1/Proto-Lbc/ Ht31 | Cardiac stress response |
| mAKAPβ | AKAP6 | AKAP100 | Excitation-contraction coupling; Cardiac stress response |
| AKAP Yotiao | AKAP9 | GC-NAP | Cardiac repolarization |
| Gravin | AKAP12 | AKAP250 | Endothelial barrier function; β-AR desensitization/resensitization cycle |
| SKIP | SPHKAP | - | Cardiac stress response |
| PDE Family | PDE Gene | Substrate Specificity | Regulated Cardiovascular Process |
|---|---|---|---|
| PDE1 | PDE1A | cAMP, cGMP | Cardiac stress response |
| PDE1B | |||
| PDE1C | |||
| PDE2 | PDE2A | cAMP, cGMP | Endothelial barrier function; Excitation-contraction coupling |
| PDE3 | PDE3A | cAMP, cGMP | Endothelial barrier function; Excitation-contraction coupling; Basal pacemaking activity of the SA node; Cardiac stress response |
| PDE3B | |||
| PDE4 | PDE4A | cAMP | Endothelial barrier function; Excitation-contraction coupling; Basal pacemaking activity of the SA node; Cardiac stress response |
| PDE4B | |||
| PDE4C | |||
| PDE4D | |||
| PDE5 | PDE5A | cGMP | Endothelial barrier function; Excitation-contraction coupling; Cardiac stress response |
| PDE8 | PDE8A | cAMP | Excitation-contraction coupling |
| PDE8B | |||
| PDE9 | PDE9A | cGMP | Cardiac stress response |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. https://doi.org/10.3390/jcdd5010014
Ercu M, Klussmann E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. Journal of Cardiovascular Development and Disease. 2018; 5(1):14. https://doi.org/10.3390/jcdd5010014
Chicago/Turabian StyleErcu, Maria, and Enno Klussmann. 2018. "Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System" Journal of Cardiovascular Development and Disease 5, no. 1: 14. https://doi.org/10.3390/jcdd5010014
APA StyleErcu, M., & Klussmann, E. (2018). Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. Journal of Cardiovascular Development and Disease, 5(1), 14. https://doi.org/10.3390/jcdd5010014