Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype

{kind=link}
{kind=link}
Abstract
1. Introduction A
1.1. Dystrophinopathies
1.2. Cardiac Phenotype of Dystrophinopathies
2. Molecular Genetics of Dystrophinopathies
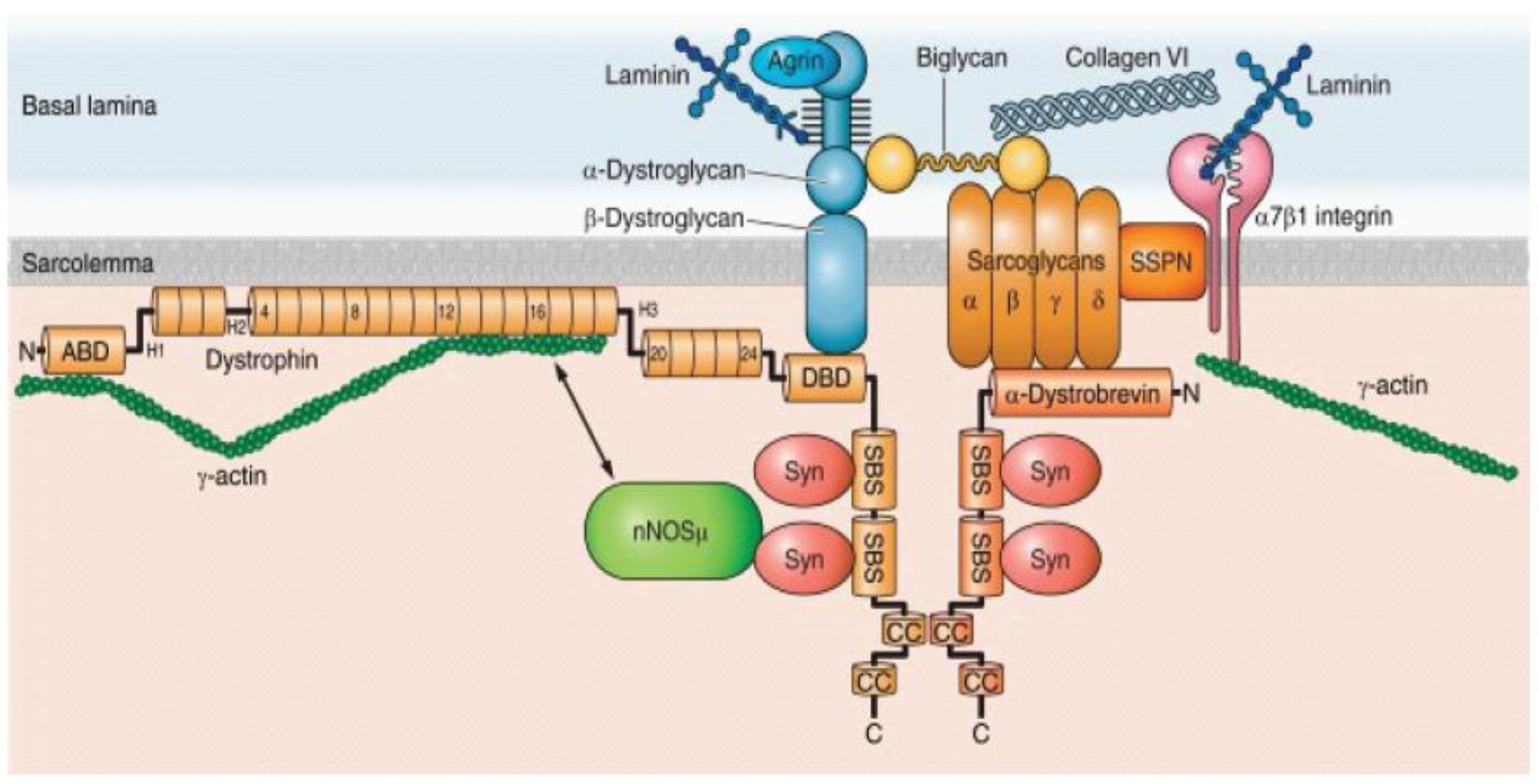
2.1. Dystrophin-Glycoproein Commplex and Its Molecular Structure
2.2. Spatial Expression of Dystrophin
2.3. Genotype-Phenotype Correlation
2.3.1. Reading Frame Rule
2.3.2. Alternative Splicing and Exon Skipping
3. Molecular Mechanisms of Dystrophic Hearts
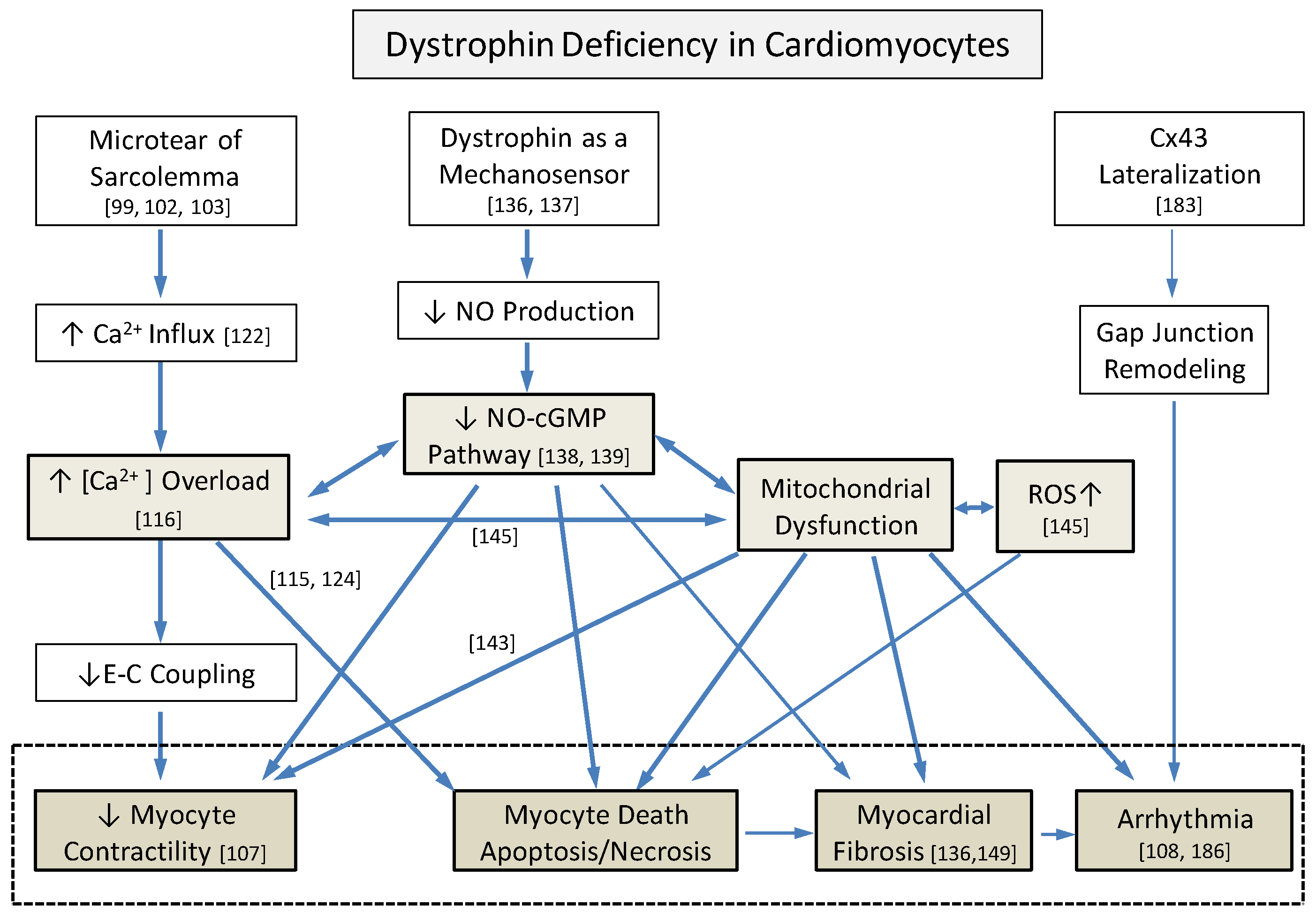
3.1. Deficiency of Structural Integrity of DGC
3.2. Secondary Abnormal Cellular Responses in Myocytes
3.2.1. Intracellular Ca2+ Increase
3.2.2. NO and nNOS Pathways
3.2.3. Increased ROS and Mitochondrial Dysfunction
3.2.4. Extracellular Matrix Remodeling and Myocardial Fibrosis
3.3. Other Associated Pathological Mechanisms
3.3.1. Epigenetic Factors
3.3.2. Post-Translational Modification
3.3.3. Telomere Dysfunction
3.3.4. Genetic Modifiers
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Wallace, G.Q.; McNally, E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Danialou, G.; Comtois, A.S.; Dudley, R.; Karpati, G.; Vincent, G.; Des Rosiers, C.; Petrof, B.J. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001, 15, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Hoogerwaard, E.M.; van der Wouw, P.A.; Wilde, A.A.; Bakker, E.; Ippel, P.F.; Oosterwijk, J.C.; Majoor-Krakauer, D.F.; van Essen, A.J.; Leschot, N.J.; de Visser, M. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 1999, 9, 347–351. [Google Scholar] [CrossRef]
- McDonald, C.M.; Abresch, R.T.; Carter, G.T.; Fowler, W.M., Jr.; Johnson, E.R.; Kilmer, D.D.; Sigford, B.J. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 1995, 74, S70–S92. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergen, J.C.; Ginjaar, H.B.; van Essen, A.J.; Pangalila, R.; de Groot, I.J.; Wijkstra, P.J.; Zijnen, M.P.; Cobben, N.A.; Kampelmacher, M.J.; Wokke, B.H.; et al. Forty-Five Years of Duchenne Muscular Dystrophy in The Netherlands. J. Neuromuscul. Dis. 2014, 1, 99–109. [Google Scholar] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Snow, W.M.; Anderson, J.E.; Jakobson, L.S. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: A review. Neurosci. Biobehav. Rev. 2013, 37, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, R.G.; Hoogland, G.; Schipper, S.; Hendriksen, J.G.; Vles, J.S.; Aalbers, M.W. A possible role of dystrophin in neuronal excitability: A review of the current literature. Neurosci. Biobehav. Rev. 2015, 51, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.J.; Molnar, G.E.; Taft, L.T. The genetic relationship of progressive muscular dystrophy (Duchenne type) and mental retardation. Dev. Med. Child Neurol. 1968, 10, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Head, S.I.; Rae, C.; Morley, J.W. Brain function in Duchenne muscular dystrophy. Brain 2002, 125, 4–13. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary cardiac issues in Duchenne muscular dystrophy. Working Group of the National Heart, Lung, and Blood Institute in collaboration with Parent Project Muscular Dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Melacini, P.; Fanin, M.; Danieli, G.A.; Fasoli, G.; Villanova, C.; Angelini, C.; Vitiello, L.; Miorelli, M.; Buja, G.F.; Mostacciuolo, M.L.; et al. Cardiac involvement in Becker muscular dystrophy. J. Am. Coll. Cardiol. 1993, 22, 1927–1934. [Google Scholar] [CrossRef]
- Saito, M.; Kawai, H.; Akaike, M.; Adachi, K.; Nishida, Y.; Saito, S. Cardiac dysfunction with Becker muscular dystrophy. Am. Heart J. 1996, 132, 642–647. [Google Scholar] [CrossRef]
- Connuck, D.M.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Lowe, A.M.; Wilkinson, J.D.; Orav, E.J.; Cuniberti, L.; Salbert, B.A.; et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 2008, 155, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stollberger, C. Cardiac involvement in Becker muscular dystrophy. Can. J. Cardiol. 2008, 24, 786–792. [Google Scholar] [CrossRef]
- Steare, S.E.; Dubowitz, V.; Benatar, A. Subclinical cardiomyopathy in Becker muscular dystrophy. Br. Heart J. 1992, 68, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.I.; Politano, L.; Limongelli, F.M.; Nigro, V.; De Rimini, M.L.; Giugliano, M.A.; Petretta, V.R.; Passamano, L.; Restucci, B.; et al. Evaluation of the cardiomyopathy in Becker muscular dystrophy. Muscle Nerve 1995, 18, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Hoogerwaard, E.M.; de Voogt, W.G.; Wilde, A.A.; van der Wouw, P.A.; Bakker, E.; van Ommen, G.J.; de Visser, M. Evolution of cardiac abnormalities in Becker muscular dystrophy over a 13-year period. J. Neurol. 1997, 244, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Fitzgerald, K.; Scavena, M.; Gidding, S.; Cox, M.O.; Marks, H.; Flanigan, K.M.; Moore, S.A. Early-progressive dilated cardiomyopathy in a family with Becker muscular dystrophy related to a novel frameshift mutation in the dystrophin gene exon 27. J. Hum. Genet. 2015, 60, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, H.; Ikeda, U.; Shimada, K.; Natsume, T.; Arahata, K. Becker muscular dystrophy with early manifestation of left heart failure. Intern. Med. 1993, 32, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Muntoni, F. Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy. Heart 2004, 90, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Diegoli, M.; Grasso, M.; Favalli, V.; Serio, A.; Gambarin, F.I.; Klersy, C.; Pasotti, M.; Agozzino, E.; Scelsi, L.; Ferlini, A.; et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J. Am. Coll. Cardiol. 2011, 58, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ii, K.; Akita, H.; Tomimatsu, H.; Kurahashi, Y.; Nakatsu, T.; Miyao, M. Clinical features and cardiopulmonary function of patients with atrophic heart in Duchenne muscular dystrophy. Jpn. Heart J. 1987, 28, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Eun, L.Y.; Choi, J.Y.; Kwon, H.E.; Lee, Y.M.; Kim, H.D.; Kang, S.W. Myocardial atrophy in children with mitochondrial disease and Duchenne muscular dystrophy. Korean J. Pediatr. 2014, 57, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Kramer, D.G.; Patel, A.R.; Maron, M.S.; Udelson, J.E. Left ventricular remodeling in heart failure: Current concepts in clinical significance and assessment. JACC Cardiovasc. Imaging 2011, 4, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Meira, Z.M.; Gurgel Giannetti, J.; da Silva, M.M.; Campos, A.F.; Barbosa Mde, M.; Starling Filho, G.M.; Ferreira Rde, A.; Zatz, M.; Rochitte, C.E. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J. Am. Coll. Cardiol. 2007, 49, 1874–1879. [Google Scholar] [CrossRef] [PubMed]
- Ramaciotti, C.; Heistein, L.C.; Coursey, M.; Lemler, M.S.; Eapen, R.S.; Iannaccone, S.T.; Scott, W.A. Left ventricular function and response to enalapril in patients with duchenne muscular dystrophy during the second decade of life. Am. J. Cardiol. 2006, 98, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Su, J.A.; Ramos-Platt, L.; Menteer, J. Left Ventricular Tonic Contraction as a Novel Biomarker of Cardiomyopathy in Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2016, 37, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Kolovou, G. Cardiac involvement in Duchenne and Becker muscular dystrophy. World J. Cardiol. 2015, 7, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Spurney, C.F. Cardiomyopathy of Duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve 2011, 44, 8–19. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Barefield, D.Y.; Puckelwartz, M.J. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015, 21, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Magalhaes, T.A.; Meira, Z.M.; Rassi, C.H.; Andrade, A.C.; Gutierrez, P.S.; Azevedo, C.F.; Gurgel-giannetti, J.; Vainzof, M.; Zatz, M.; et al. Myocardial Fibrosis Progression in Duchenne and Becker Muscular Dystrophy: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.Y.; Allen, H.D.; Kim, J.J.; Valdes, S.O.; Wang, Y.; Pignatelli, R.H.; Lotze, T.E.; Miyake, C.Y. Relation of Cardiac Dysfunction to Rhythm Abnormalities in Patients with Duchenne or Becker Muscular Dystrophies. Am. J. Cardiol. 2016, 117, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.O.; Morgan, T.M.; Burnette, W.B.; Markham, L.W. Correlation of heart rate and cardiac dysfunction in Duchenne muscular dystrophy. Pediatr. Cardiol. 2012, 33, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; D’Orsogna, L.; O’Shea, J.P. Autonomic function and the sinus tachycardia of Duchenne muscular dystrophy. Brain Dev. 1989, 11, 247–250. [Google Scholar] [CrossRef]
- Lanza, G.A.; Dello Russo, A.; Giglio, V.; De Luca, L.; Messano, L.; Santini, C.; Ricci, E.; Damiani, A.; Fumagalli, G.; De Martino, G.; et al. Impairment of cardiac autonomic function in patients with Duchenne muscular dystrophy: Relationship to myocardial and respiratory function. Am. Heart J. 2001, 141, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Kedem, J.; Sonn, J.; Scheinowitz, M.; Weiss, H.R. Relationship between local oxygen consumption and local and external cardiac work: Effect of tachycardia. Cardiovasc. Res. 1989, 23, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Van Ruiten, H.J.; Marini Bettolo, C.; Cheetham, T.; Eagle, M.; Lochmuller, H.; Straub, V.; Bushby, K.; Guglieri, M. Why are some patients with Duchenne muscular dystrophy dying young: An analysis of causes of death in North East England. Eur. J. Paediatr. Neurol. 2016, 20, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Groh, W.J. Arrhythmias in the muscular dystrophies. Heart Rhythm. 2012, 9, 1890–1895. [Google Scholar] [CrossRef] [PubMed]
- Bies, R.D.; Phelps, S.F.; Cortez, M.D.; Roberts, R.; Caskey, C.T.; Chamberlain, J.S. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992, 20, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Torelli, S.; Ferlini, A.; Obici, L.; Sewry, C.; Muntoni, F. Expression, regulation and localisation of dystrophin isoforms in human foetal skeletal and cardiac muscle. Neuromuscul. Disord. 1999, 9, 541–551. [Google Scholar] [CrossRef]
- Tinsley, J.M.; Blake, D.J.; Pearce, M.; Knight, A.E.; Kendrick-Jones, J.; Davies, K.E. Dystrophin and related proteins. Curr. Opin. Genet. Dev. 1993, 3, 484–490. [Google Scholar] [CrossRef]
- Anastasi, G.; Cutroneo, G.; Gaeta, R.; Di Mauro, D.; Arco, A.; Consolo, A.; Santoro, G.; Trimarchi, F.; Favaloro, A. Dystrophin-glycoprotein complex and vinculin-talin-integrin system in human adult cardiac muscle. Int. J. Mol. Med. 2009, 23, 149–159. [Google Scholar] [PubMed]
- Iwata, Y.; Pan, Y.; Hanada, H.; Yoshida, T.; Shigekawa, M. Dystrophin-glycoprotein complex purified from hamster cardiac muscle. Comparison of the complexes from cardiac and skeletal muscles of hamster and rabbit. J. Mol. Cell. Cardiol. 1996, 28, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- Klietsch, R.; Ervasti, J.M.; Arnold, W.; Campbell, K.P.; Jorgensen, A.O. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ. Res. 1993, 72, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Dent, K.M.; Dunn, D.M.; von Niederhausern, A.C.; Aoyagi, A.T.; Kerr, L.; Bromberg, M.B.; Hart, K.J.; Tuohy, T.; White, S.; den Dunnen, J.T.; et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am. J. Med. Genet. A 2005, 134, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Gappmaier, E.; Howard, M.T.; Sampson, J.; Mendell, J.; Wall, C.; King, W.; et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum. Mutat. 2009, 30, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Buzin, C.H.; Feng, J.; Yan, J.; Serrano, C.; Sangani, D.S.; Wall, C.; Prior, T.W.; Sommer, S.S. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001, 57, 645–650. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Aartsma-Rus, A.; Flanigan, K.M.; Weiss, R.B.; Kneppers, A.L.; Lalic, T.; Janson, A.A.; Ginjaar, H.B.; Breuning, M.H.; den Dunnen, J.T. Duplications in the DMD gene. Hum. Mutat. 2006, 27, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Nachman, M.W.; Crowell, S.L. Contrasting evolutionary histories of two introns of the duchenne muscular dystrophy gene, Dmd, in humans. Genetics 2000, 155, 1855–1864. [Google Scholar] [PubMed]
- Ahn, A.H.; Kunkel, L.M. The structural and functional diversity of dystrophin. Nat. Genet. 1993, 3, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Kaprielian, R.R.; Stevenson, S.; Rothery, S.M.; Cullen, M.J.; Severs, N.J. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 2000, 101, 2586–2594. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, N.; Shidoh, Y.; Kondo, S.; Takatoh, J.; Hanaoka, K. Subcellular localization of dystrophin isoforms in cardiomyocytes and phenotypic analysis of dystrophin-deficient mice reveal cardiac myopathy is predominantly caused by a deficiency in full-length dystrophin. Exp. Anim. 2013, 62, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F. Brain dystrophin, neurogenetics and mental retardation. Brain Res. Brain Res. Rev. 2000, 32, 277–307. [Google Scholar] [CrossRef]
- Chelly, J.; Hamard, G.; Koulakoff, A.; Kaplan, J.C.; Kahn, A.; Berwald-Netter, Y. Dystrophin gene transcribed from different promoters in neuronal and glial cells. Nature 1990, 344, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Perronnet, C.; Vaillend, C. Dystrophins, utrophins, and associated scaffolding complexes: Role in mammalian brain and implications for therapeutic strategies. J. Biomed. Biotechnol. 2010, 2010, 849426. [Google Scholar] [PubMed]
- Tinsley, J.M.; Davies, K.E. Utrophin: A potential replacement for dystrophin? Neuromuscul. Disord. 1993, 3, 537–539. [Google Scholar] [CrossRef]
- Ohlendieck, K.; Ervasti, J.M.; Matsumura, K.; Kahl, S.D.; Leveille, C.J.; Campbell, K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron 1991, 7, 499–508. [Google Scholar] [CrossRef]
- Blake, D.J.; Tinsley, J.M.; Davies, K.E. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 1996, 6, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Selig, S.; Kunkel, L.M. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum. Mol. Genet. 1995, 4, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Pillers, D.A.; Bulman, D.E.; Weleber, R.G.; Sigesmund, D.A.; Musarella, M.A.; Powell, B.R.; Murphey, W.H.; Westall, C.; Panton, C.; Becker, L.E.; et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat. Genet. 1993, 4, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Kesari, A.; Pirra, L.N.; Bremadesam, L.; McIntyre, O.; Gordon, E.; Dubrovsky, A.L.; Viswanathan, V.; Hoffman, E.P. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum. Mutat. 2008, 29, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A. X-Linked Dilated Cardiomyopathy: A Cardiospecific Phenotype of Dystrophinopathy. Pharmaceuticals 2015, 8, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Beggs, A.H. Dystrophinopathy, the expanding phenotype. Dystrophin abnormalities in X-linked dilated cardiomyopathy. Circulation 1997, 95, 2344–2347. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Nakamura, A.; Yazaki, M.; Ikeda, S.; Takeda, S. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1998, 7, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Melis, M.A.; Ganau, A.; Dubowitz, V. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X-linked dilated cardiomyopathy. Am. J. Hum. Genet. 1995, 56, 151–157. [Google Scholar] [PubMed]
- Muntoni, F.; Wilson, L.; Marrosu, G.; Marrosu, M.G.; Cianchetti, C.; Mestroni, L.; Ganau, A.; Dubowitz, V.; Sewry, C. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J. Clin. Investig. 1995, 96, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Franz, W.M.; Muller, M.; Muller, O.J.; Herrmann, R.; Rothmann, T.; Cremer, M.; Voit, T.; Rothmann, T.; Cremer, M. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X-linked dilated cardiomyopathy. Lancet 2000, 355, 1781–1785. [Google Scholar] [CrossRef]
- Singh, S.M.; Bandi, S.; Shah, D.D.; Armstrong, G.; Mallela, K.M. Missense mutation Lys18Asn in dystrophin that triggers X-linked dilated cardiomyopathy decreases protein stability, increases protein unfolding, and perturbs protein structure, but does not affect protein function. PLoS ONE 2014, 9, e110439. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, S.B.; Sherratt, T.G.; Heckmatt, J.Z.; Dubowitz, V.; Miller, G.; Shokeir, M.; Ray, P.N.; Strong, P.N.; Worton, R.G. Dystrophin in frameshift deletion patients with Becker muscular dystrophy. Am. J. Hum. Genet. 1992, 51, 562–570. [Google Scholar] [PubMed]
- Winnard, A.V.; Mendell, J.R.; Prior, T.W.; Florence, J.; Burghes, A.H. Frameshift deletions of exons 3–7 and revertant fibers in Duchenne muscular dystrophy: Mechanisms of dystrophin production. Am. J. Hum. Genet. 1995, 56, 158–166. [Google Scholar] [PubMed]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Howard, M.T.; Sampson, J.B.; Swoboda, K.J.; Bromber, M.B.; Mendal, J.R.; Taylor, L.E.; et al. Nonsense mutation-associated Becker muscular dystrophy: Interplay between exon definition and splicing regulatory elements within the DMD gene. Hum. Mutat. 2011, 32, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Dunn, D.; Larsen, C.A.; Medne, L.; Bonnemann, C.B.; Weiss, R.B. Becker muscular dystrophy due to an inversion of exons 23 and 24 of the DMD gene. Muscle Nerve 2011, 44, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Winnard, A.V.; Klein, C.J.; Coovert, D.D.; Prior, T.; Papp, A.; Snyder, P.; Bulman, D.E.; Ray, P.N.; McAndrew, P.; King, W.; et al. Characterization of translational frame exception patients in Duchenne/Becker muscular dystrophy. Hum. Mol. Genet. 1993, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Freda, M.P.; Vitiello, L.; Danieli, G.A.; Pegoraro, E.; Angelini, C. Duchenne phenotype with in-frame deletion removing major portion of dystrophin rod: Threshold effect for deletion size? Muscle Nerve 1996, 19, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Gobbi, P.; Sewry, C.; Sherratt, T.; Taylor, J.; Sandhu, S.K.; Abbs, S.; Roberts, R.; Hodgson, S.V.; Bobrow, M. Deletions in the 5’ region of dystrophin and resulting phenotypes. J. Med. Genet. 1994, 31, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.V.; Johnson, M.A.; Bushby, K.M.; Gardner-Medwin, D.; Curtis, A.; Ginjaar, I.B.; den Dunnen, J.T.; Welch, J.L.; Butler, T.J.; Bakker, E. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 1. Trends across the clinical groups. J. Med. Genet. 1993, 30, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Vainzof, M.; Takata, R.I.; Passos-Bueno, M.R.; Pavanello, R.C.; Zatz, M. Is the maintainance of the C-terminus domain of dystrophin enough to ensure a milder Becker muscular dystrophy phenotype? Hum. Mol. Genet. 1993, 2, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Love, D.R.; Flint, T.J.; Genet, S.A.; Middleton-Price, H.R.; Davies, K.E. Becker muscular dystrophy patient with a large intragenic dystrophin deletion: Implications for functional minigenes and gene therapy. J. Med. Genet. 1991, 28, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Tuffery-Giraud, S.; Beroud, C.; Leturcq, F.; Yaou, R.B.; Hamroun, D.; Michel-Calemard, L.; Michel-Calemard, L.; Moizard, M.P.; Bernard, R.; Cossee, M. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase. Hum. Mutat. 2009, 30, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Peddareddygari, L.R.; Pillai, B.H.; Nochlin, D.; Sharer, L.R.; Grewal, R.P. Phenotype-genotype analysis of dystrophinopathy caused by duplication mutation in Dystrophin gene in an African patient. Afr. Health Sci. 2011, 11, 607–609. [Google Scholar] [PubMed]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, p. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef] [PubMed]
- Im, W.B.; Phelps, S.F.; Copen, E.H.; Adams, E.G.; Slightom, J.L.; Chamberlain, J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996, 5, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Janssen, P.M.; Hiranandani, N.; Mays, T.A.; Rafael-Fortney, J.A. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2373–H2378. [Google Scholar] [CrossRef] [PubMed]
- Megeney, L.A.; Kablar, B.; Perry, R.L.; Ying, C.; May, L.; Rudnicki, M.A. Severe cardiomyopathy in mice lacking dystrophin and MyoD. Proc. Natl. Acad. Sci. USA 1999, 96, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Van Westering, T.L.; Betts, C.A.; Wood, M.J. Current Understanding of Molecular Pathology and Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.; Yasuda, S.; Chamberlain, J.; Metzger, J.M. Cardiac consequences to skeletal muscle-centric therapeutics for Duchenne muscular dystrophy. Trends Cardiovasc. Med. 2009, 19, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, T.G. Epigenetic Regulation in Heart Failure: Part I RNA. Cardiol. Rev. 2015, 23, 213–228. [Google Scholar] [CrossRef] [PubMed]
- DiSalvo, T.G. Epigenetic regulation in heart failure: Part II DNA and chromatin. Cardiol. Rev. 2015, 23, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Mourkioti, F.; Kustan, J.; Kraft, P.; Day, J.W.; Zhao, M.M.; Kost-Alimova, M.; Protopopov, A.; DePinho, R.A.; Bernstein, D.; Meeker, A.K.; et al. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol. 2013, 15, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Ong, S.G.; LaGory, E.L.; Kraft, P.E.; Giaccia, A.J.; Wu, J.C.; Blau, H.M. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2016, 113, 13120–13125. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Rosati, J.; Straino, S.; Spallotta, F.; Berni, R.; Stilli, D.; Rossic, S.; Mussoc, E.; Macchic, E.; Mai, A.; et al. Nepsilon-lysine acetylation determines dissociation from GAP junctions and lateralization of connexin 43 in normal and dystrophic heart. Proc. Natl. Acad. Sci. USA 2011, 108, 2795–2800. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Polakova, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Dowling, P.; Zweyer, M.; Mundegar, R.R.; Henry, M.; Meleady, P.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J. Proteom. 2016, 145, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy. Biomed. Res. Int. 2014, 2014, 246195. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, M.; Zeiger, U.; Forderer, M.; Brinkmeier, H.; Fink, R.H. Cardiac and respiratory dysfunction in Duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 2013, 33, 1174–1213. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000, 150, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T.; et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Martins, A.S.; Niggli, E.; Shirokova, N. Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008, 77, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; Lopez de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Gorski, P.A.; Ceholski, D.K.; Hajjar, R.J. Altered myocardial calcium cycling and energetics in heart failure—A rational approach for disease treatment. Cell Metab. 2015, 21, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.Y.; Turner, P.R.; Denetclaw, W.F.; Steinhardt, R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 1990, 250, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Franco, A., Jr.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009, 46, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on (Ca2+)i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Adams, A.M.; Davies, S.M.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Schaus, B.J.; Fallavollita, J.A.; Lee, T.C.; Canty, J.M., Jr. Preload induces troponin I degradation independently of myocardial ischemia. Circulation 2001, 103, 2035–2037. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.L.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B. Role of nitric oxide in skeletal muscle: Synthesis, distribution and functional importance. Acta. Physiol. Scand. 1998, 162, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Meissner, G. Physiology of nitric oxide in skeletal muscle. Physiol. Rev. 2001, 81, 209–237. [Google Scholar] [PubMed]
- Xu, K.Y.; Kuppusamy, S.P.; Wang, J.Q.; Li, H.; Cui, H.; Dawson, T.M.; Huang, P.L.; Burnett, A.L.; Kuppusamy, P.; Becker, L.C. Nitric oxide protects cardiac sarcolemmal membrane enzyme function and ion active transport against ischemia-induced inactivation. J. Biol. Chem. 2003, 278, 41798–41803. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Huso, D.L.; Dawson, T.M.; Bredt, D.S.; Becker, L.C. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1999, 96, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Kanai, A.J.; Pearce, L.L.; Clemens, P.R.; Birder, L.A.; VanBibber, M.M.; Choi, S.Y.; de Groat, W.C.; Peterson, J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc. Natl. Acad. Sci. USA 2001, 98, 14126–14131. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; Crassous, P.A.; Schneider, J.S.; Beuve, A.; Fraidenraich, D. Neuronal nitric oxide synthase localizes to utrophin expressing intercalated discs and stabilizes their structural integrity. Neuromuscul. Disord. 2015, 25, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.A.; Zheng, R.; Gonzalez, J.P.; Xie, L.H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric oxide signalling pathway in Duchenne muscular dystrophy mice: Up-regulation of L-arginine transporters. Biochem. J. 2013, 449, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Zhang, L.; Adams, M.E.; Phillips, A.; Freitas, M.A.; Froehner, S.C.; Green-Church, K.B.; Montanaro, F. Proteomic analysis reveals new cardiac-specific dystrophin-associated proteins. PLoS ONE 2012, 7, e43515. [Google Scholar] [CrossRef] [PubMed]
- Wehling-Henricks, M.; Jordan, M.C.; Roos, K.P.; Deng, B.; Tidball, J.G. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 2005, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Michele, D.E. Dystrophin-glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.M.; Dai, D.F.; Percival, J.M.; Minami, E.; Willis, M.S.; Patrucco, E.; Froehnerc, S.C.; Beavoa, J.A. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 19079–19083. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.J.; Young, M.E.; Allen, B.G.; Gillis, M.A.; Danialou, G.; Deschepper, C.F.; Petrof, B.J.; Des Rosiers, C. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 7028–7033. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.Z.; Reiter, M.; Gastonguay, C.; McGivern, J.V.; Guan, X.; Ge, Z.D.; Mack, D.L.; Childers, M.K.; Ebert, A.D.; Strande, J.L. Nicorandil, a Nitric Oxide Donor and ATP-Sensitive Potassium Channel Opener, Protects Against Dystrophin-Deficient Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Gurtner, A.; Rosati, J.; Illi, B.; Ragone, G.; Piaggio, G.; Moggio, M.; Lamperti, C.; D'Angelo, G.; Clementi, E.; et al. Nitric oxide deficiency determines global chromatin changes in Duchenne muscular dystrophy. FASEB J. 2009, 23, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.L.; Heymes, D.C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Alonso, M.T.; Villalobos, C.; Chamero, P.; Alvarez, J.; Garcia-Sancho, J. Calcium microdomains in mitochondria and nucleus. Cell Calcium. 2006, 40, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.A.; Allen, D.G. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1969–H1977. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, M.; Khairallah, R.; Young, M.E.; Dyck, J.R.; Petrof, B.J.; Des Rosiers, C. Metabolic and signaling alterations in dystrophin-deficient hearts precede overt cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell. Physiol. Biochem. 2007, 20, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Papavasiliou, A.; Spargias, K.; Constandoulakis, P.; Papadopoulos, G.; Karanasios, E.; Georgakopoulos, D.; Kolovou, G.; Demerouti, E.; Polymeros, S.; et al. Myocardial inflammation in Duchenne Muscular Dystrophy as a precipitating factor for heart failure: A prospective study. BMC Neurol. 2010, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Delfin, D.A.; Zang, K.E.; Schill, K.E.; Patel, N.T.; Janssen, P.M.; Raman, S.V.; Rafael-Fortney, J.A. Cardiomyopathy in the dystrophin/utrophin-deficient mouse model of severe muscular dystrophy is characterized by dysregulation of matrix metalloproteinases. Neuromuscul. Disord. 2012, 22, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Delfin, D.A.; Xu, Y.; Peterson, J.M.; Guttridge, D.C.; Rafael-Fortney, J.A.; Janssen, P.M. Improvement of cardiac contractile function by peptide-based inhibition of NF-kappaB in the utrophin/dystrophin-deficient murine model of muscular dystrophy. J. Transl. Med. 2011, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Ameen, V.; Robson, L.G. Experimental models of duchenne muscular dystrophy: Relationship with cardiovascular disease. Open Cardiovasc. Med. J. 2010, 4, 265–277. [Google Scholar] [PubMed]
- Bridges, L.R. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J. Neurol. Sci. 1986, 72, 147–157. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Dai, H.; Chiba, Y.; Uematsu, M.; Hino-Fukuyo, N.; Onuma, A.; Iinuma, K.; Tsuchiya, S. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J. Neurol. Sci. 2009, 280, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, P.; Bernasconi, P.; Cornelio, F.; Mantegazza, R. Transforming growth factor-beta 1 in polymyositis and dermatomyositis correlates with fibrosis but not with mononuclear cell infiltrate. J. Neuropathol. Exp. Neurol. 1997, 56, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Haginoya, K.; Wu, Y.; Chiba, Y.; Nakanishi, T.; Onuma, A.; Sato, Y.; Takigawa, M.; Iinuma, K.; Tsuchiya, S. Connective tissue growth factor is overexpressed in muscles of human muscular dystrophy. J. Neurol. Sci. 2008, 267, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Au, C.G.; Butler, T.L.; Sherwood, M.C.; Egan, J.R.; North, K.N.; Winlaw, D.S. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int. J. Exp. Pathol. 2011, 92, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Bizzarro, V.; Petrella, A.; Parente, L. Annexin A1: Novel roles in skeletal muscle biology. J. Cell. Physiol. 2012, 227, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Camors, E.; Monceau, V.; Charlemagne, D. Annexins and Ca2+ handling in the heart. Cardiovasc. Res. 2005, 65, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Han, P. Epigenetic and lncRNA regulation of cardiac pathophysiology. Biochim. Biophys Acta 2016, 1863, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, W.W.; Hu, S.J. Heart failure: Advanced development in genetics and epigenetics. Biomed. Res. Int. 2015, 2015, 352734. [Google Scholar] [CrossRef] [PubMed]
- Piran, S.; Liu, P.; Morales, A.; Hershberger, R.E. Where genome meets phenome: Rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J. Am. Coll. Cardiol. 2012, 60, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.A.; Poizat, C. Epigenetics and chromatin remodeling in adult cardiomyopathy. J. Pathol. 2013, 231, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Lamar, K.M.; McNally, E.M. Genetic Modifiers for Neuromuscular Diseases. J. Neuromuscul. Dis. 2014, 1, 3–13. [Google Scholar] [PubMed]
- Vo, A.H.; McNally, E.M. Modifier genes and their effect on Duchenne muscular dystrophy. Curr. Opin. Neurol. 2015, 28, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Nanni, S.; Re, A.; Ripoli, C.; Gowran, A.; Nigro, P.; D’Amario, D.; Amodeo, A.; Crea, F.; Grassi, C.; Pontecorvi, A.; et al. The nuclear pore protein Nup153 associates with chromatin and regulates cardiac gene expression in dystrophic mdx hearts. Cardiovasc. Res. 2016, 112, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Kalverda, B.; Pickersgill, H.; Shloma, V.V.; Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 2010, 140, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; De Simone, M.; Colussi, C.; Zaccagnini, G.; Fasanaro, P.; Pescatori, M.; Cardani, R.; Perbellini, R.; Isaia, E.; Sale, P.; et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J. 2009, 23, 3335–3346. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar] [CrossRef] [PubMed]
- Cassano, M.; Berardi, E.; Crippa, S.; Toelen, J.; Barthelemy, I.; Micheletti, R.; Chuah, M.; Vandendriessche, T.; Debyser, Z.; Blot, S.; et al. Alteration of cardiac progenitor cell potency in GRMD dogs. Cell Transplant. 2012, 21, 1945–1967. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal role of miR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Florian, A.; Patrascu, A.; Rosch, S.; Waltenberger, J.; Sechtem, U.; Schwab, M.; Schaeffeler, E.; Yilmaz, A. Identification of cardiomyopathy associated circulating miRNA biomarkers in patients with muscular dystrophy using a complementary cardiovascular magnetic resonance and plasma profiling approach. J. Cardiovasc. Magn. Reson. 2016, 18, 25. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Kieken, F.; Mutsaers, N.; Dolmatova, E.; Virgil, K.; Wit, A.L.; Kellezi, A.; Hirst-Jensen, B.J.; Duffy, H.S.; Sorgen, P.L. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 2009, 104, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.S.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys Acta 2012, 1818, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; Ramachandran, J.; Xie, L.H.; Contreras, J.E.; Fraidenraich, D. Selective Connexin43 Inhibition Prevents Isoproterenol-Induced Arrhythmias and Lethality in Muscular Dystrophy Mice. Sci. Rep. 2015, 5, 13490. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [PubMed]
- Vetrone, S.A.; Montecino-Rodriguez, E.; Kudryashova, E.; Kramerova, I.; Hoffman, E.P.; Liu, S.D.; Miceli, M.C.; Spencer, M.J. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J. Clin. Investig. 2009, 119, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Ceco, E.; Lamar, K.M.; Kaminoh, Y.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol. 2013, 73, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Barp, A.; Bello, L.; Politano, L.; Melacini, P.; Calore, C.; Polo, A.; Vianello, S.; Soraru, G.; Semplicini, C.; Pantic, B.; et al. Genetic Modifiers of Duchenne Muscular Dystrophy and Dilated Cardiomyopathy. PLoS ONE 2015, 10, e0141240. [Google Scholar] [CrossRef] [PubMed]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [PubMed]
- Beytia Mde, L.; Vry, J.; Kirschner, J. Drug treatment of Duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol. 2012, 31, 4–8. [Google Scholar] [PubMed]
- Rodino-Klapac, L.R.; Mendell, J.R.; Sahenk, Z. Update on the treatment of Duchenne muscular dystrophy. Curr. Neurol. Neurosci. Rep. 2013, 13, 332. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuda, T.; Fitzgerald, K.K. Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype. J. Cardiovasc. Dev. Dis. 2017, 4, 14. https://doi.org/10.3390/jcdd4030014
Tsuda T, Fitzgerald KK. Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype. Journal of Cardiovascular Development and Disease. 2017; 4(3):14. https://doi.org/10.3390/jcdd4030014
Chicago/Turabian StyleTsuda, Takeshi, and Kristi K. Fitzgerald. 2017. "Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype" Journal of Cardiovascular Development and Disease 4, no. 3: 14. https://doi.org/10.3390/jcdd4030014
APA StyleTsuda, T., & Fitzgerald, K. K. (2017). Dystrophic Cardiomyopathy: Complex Pathobiological Processes to Generate Clinical Phenotype. Journal of Cardiovascular Development and Disease, 4(3), 14. https://doi.org/10.3390/jcdd4030014