Midkine’s Role in Cardiac Pathology


Abstract
1. Introduction
2. MDK in Normal and Pathophysiology
3. Circulating MDK
3.1. Receptor-Type Protein Tyrosine Phosphatase ζ (RTPTPζ)
3.2. Integrins
3.3. Low-Density Lipoprotein (LDL) Receptor-Related Protein (LRP)
3.4. Notch
3.5. Other
4. Intracellular MDK
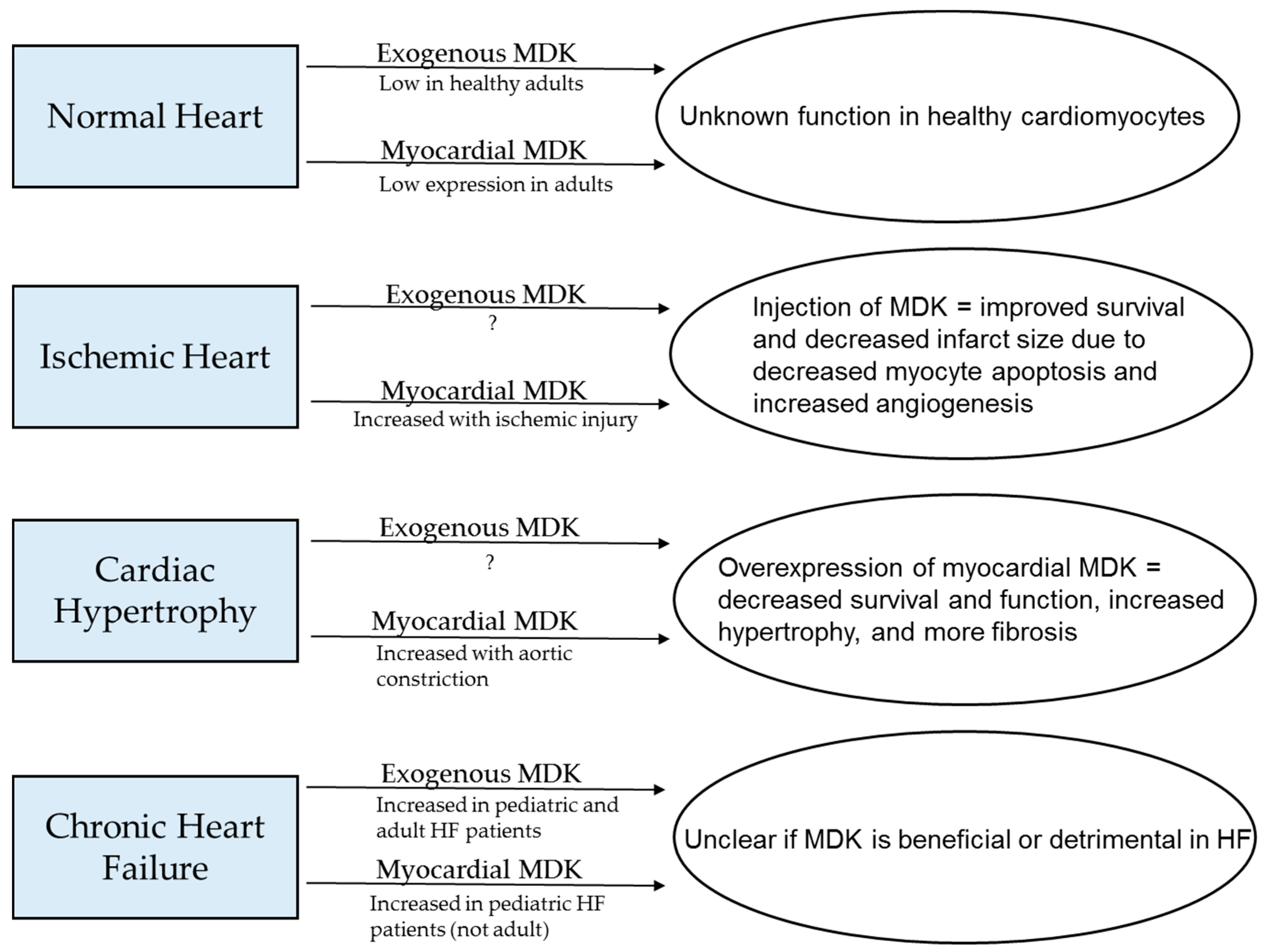
5. MDK in Cardiac Pathology
5.1. Phenotype of Genetically Manipulated Models
5.2. Models of Cardiac Pathology and MDK
5.2.1. Ischemia/Reperfusion Injury
5.2.2. Myocardial Infarction
5.2.3. Thoracic Aortic Constriction
5.2.4. Continuous Pacing
5.2.5. Indirect Cardiac Pathology
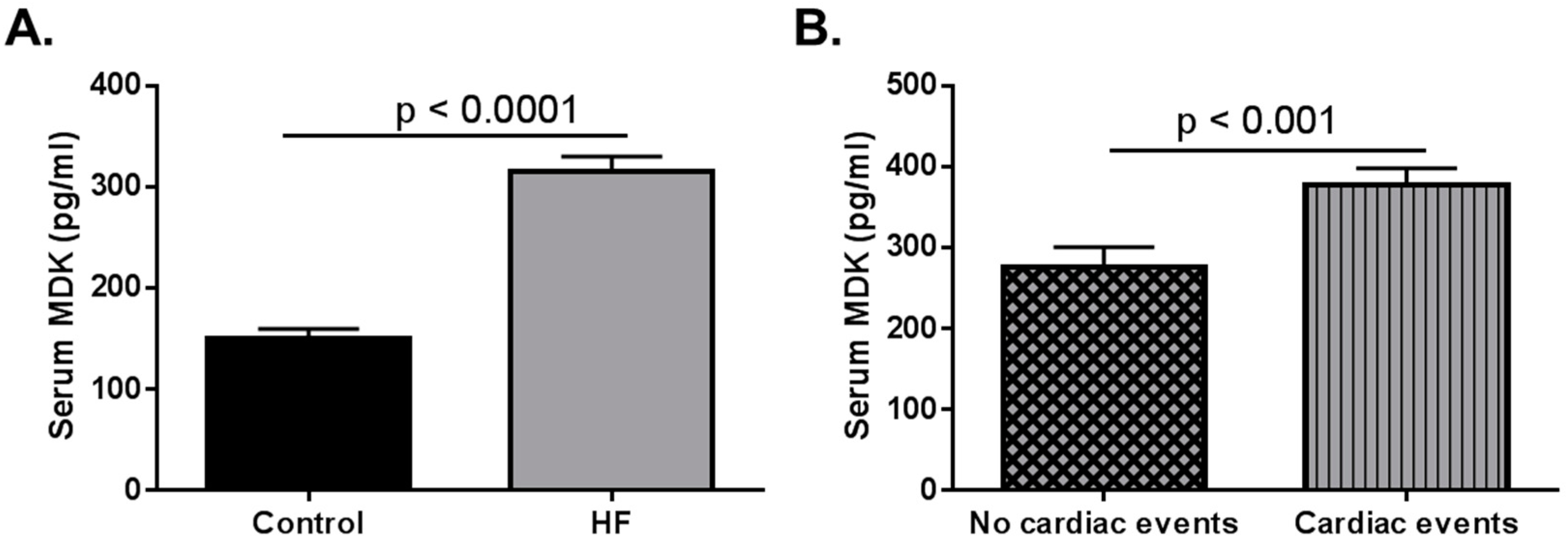
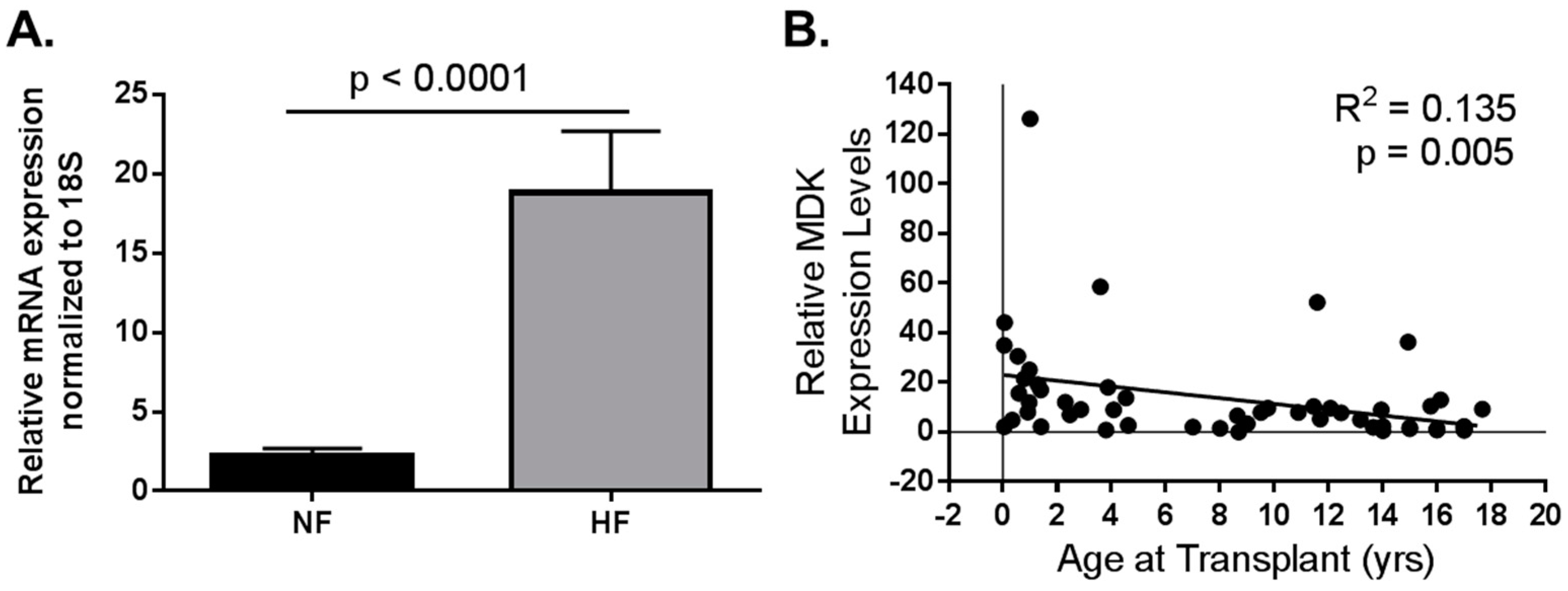
5.3. Consideration for Pediatric HF
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kadomatsu, K. Midkine, a heparin-binding growth factor: Its discovery and functions. J. Jpn. Biochem. Soc. 1998, 70, 1315–1325. [Google Scholar]
- Tomomura, M.; Kadomatsu, K.; Nakamoto, M.; Muramatsu, H.; Kondoh, H.; Imagawa, K.-I.; Muramatsu, T. A retinoic acid responsive gene, MK, produces a secreted protein with heparin binding activity. Biochem. Biophys. Res. Commun. 1990, 171, 603–609. [Google Scholar] [CrossRef]
- Nakamoto, M.; Matsubara, S.; Miyauchi, T.; Obama, H.; Ozawa, M.; Muramatsu, T. A new family of heparin binding growth/differentiation factors: Differential expression of the midkine (MK) and HB-GAM genes during mouse development. J. Biochem. 1992, 112, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Uehara, K.; Matsubara, S.; Kadomatsu, K.; Tsutsui, J.-I.; Muramatsu, T. Genomic structure of human midkine (MK), a retinoic acid-responsive growth/differentiation factor. J. Biochem. 1992, 111, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Shirahama, H.; Yonezawa, S.; Maruta, H.; Muramatsu, T. Midkine, a retinoic acid-inducible growth/differentiation factor: Immunochemical evidence for the function and distribution. Dev. Biol. 1993, 159, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Midkine (MK), the product of a retinoic acid responsive gene, and pleiotrophin constitute a new protein family regulating growth and differentiation. Int. J. Dev. Biol. 1993, 37, 183–188. [Google Scholar] [PubMed]
- Satoh, J.; Muramatsu, H.; Moretto, G.; Muramatsu, T.; Chang, H.J.; Kim, S.T.; Cho, J.M.; Kim, S.U. Midkine that promotes survival of fetal human neurons is produced by fetal human astrocytes in culture. Dev. Brain Res. 1993, 75, 201–205. [Google Scholar] [CrossRef]
- Xu, C.; Zhu, S.; Wu, M.; Han, W.; Yu, Y. Functional receptors and intracellular signal pathways of midkine (MK) and pleiotrophin (PTN). Biol. Pharm. Bull. 2014, 37, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Badila, E.; Daraban, A.M.; Ţintea, E.; Bartoş, D.; Alexandru, N.; Georgescu, A. Midkine proteins in cardio-vascular disease. Where do we come from and where are we heading to? Eur. J. Pharmacol. 2015, 762, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Structure and function of midkine as the basis of its pharmacological effects. Br. J. Pharmacol. 2014, 171, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Kishida, S.; Tsubota, S. The heparin-binding growth factor midkine: The biological activities and candidate receptors. J. Biochem. 2013, 153, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.R. Measuring midkine: The utility of midkine as a biomarker in cancer and other diseases. Br. J. Pharmacol. 2014, 171, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Bencsik, P.; Görbe, A.; Csonka, C.; Sakamoto, K.; Kishida, S.; Ferdinandy, P. Therapeutic potential of midkine in cardiovascular disease. Br. J. Pharmacol. 2014, 171, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Bu, G.; Chen, S.; Takei, Y.; Hibi, K.; Kodera, Y.; McCormick, Y.M.; Nakao, A.; Noda, M.; Muramatsu, T.; et al. Premature ligand-receptor interaction during biosynthesis limits the production of growth factor midkine and its receptor LDL receptor-related protein 1. J. Biol. Chem. 2011, 286, 8405–8413. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Tomomura, M.; Kadomatsu, K.; Muramatsu, T. Structure of a retinoic acid-responsive gene, MK, which is transiently activated during the differentiation of embryonal carcinoma cells and the mid-gestation period of mouse embryogenesis. J. Biol. Chem. 1990, 265, 9441–9443. [Google Scholar] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/gene/4192 (accssed on 15 June 2017).
- Pedraza, C.; Matsubara, S.; Muramatsu, T. A retinoic acid-responsive element in human midkine gene. J. Biochem. 1995, 117, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Mucenski, M.L.; Cras, T.D.L.; Nichols, W.C.; Whitsett, J.A. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J. Biol. Chem. 2004, 279, 37124–37132. [Google Scholar] [CrossRef] [PubMed]
- Takada, J.; Ooboshi, H.; Ago, T.; Kitazono, T.; Yao, H. Postischemic gene transfer of midkine, a neurotrophic factor, protects against focal brain ischemia. Gene Ther. 2005, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yuan, Y.; Lin, S.; Chang, Y.; Zhuo, X.; Wei, W.; Tao, P.; Ruan, L.; Li, Q.; Li, Z. Transiently truncated and differentially regulated expression of midkine during mouse embryogenesis. Biochem. Biophys. Res. Commun. 2005, 330, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, I.; Kaname, T.; Nakayama, T.; Nakamori, S.; Yagyu, T.; Monden, T.; Kikkawa, N.; Nishisho, I.; Muramatsu, T.; Monden, M.; et al. Expression of truncated midkine in human colorectal cancers. Cancer Lett. 1996, 106, 287–291. [Google Scholar] [CrossRef]
- Iwasaki, W.; Nagata, K.; Hatanaka, H.; Inui, T.; Kimura, T.; Muramatsu, T.; Yoshida, K.; Tasumi, M.; Inagaki, F. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997, 16, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Herradon, G.; Ezquerra, L.; Nguyen, T.; Silos-Santiago, I.; Deuel, T.F. Midkine regulates pleiotrophin organ-specific gene expression: Evidence for transcriptional regulation and functional redundancy within the pleiotrophin/midkine developmental gene family. Biochem. Biophys. Res. Commun. 2005, 333, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Mitsiadis, T.A.; Muramatsu, T.; Muramatsu, H.; Thesleff, I. Midkine (MK), a heparin-binding growth/differentiation factor, is regulated by retinoic acid and epithelial-mesenchymal interactions in the developing mouse tooth, and affects cell proliferation and morphogenesis. J. Cell Biol. 1995, 129, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Shinozawa, T.; Kato, S.; Terada, T. Divergent expression of midkine in the human fetal liver and kidney: Immunohistochemical analysis of developmental changes in hilar primitive bile ducts and hepatocytes. Liver 2000, 20, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, I.; Kaname, T.; Shin, E.; Wakasugi, E.; Monden, T.; Takatsuka, Y.; Kikkawa, N.; Muramatsu, T.; Monden, M.; Akiyama, T. Midkine expression in human breast cancers: Expression of truncated form. Breast Cancer Res. Treat. 1997, 43, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cao, G.; Wang, H.; Wang, Q.; Hou, Y. The expression and location of midkine in gastric carcinomas of Chinese patients. Cell Mol. Immunol. 2007, 4, 135–140. [Google Scholar] [PubMed]
- Muramatsu, T. Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 410–425. [Google Scholar] [CrossRef] [PubMed]
- Salaru, D.L.; Mertens, P.R. Lessons from the heart and ischemic limbs: Midkine as anti-inflammatory mediator for kidney diseases? Int. Urol. Nephrol. 2013, 45, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Muramatsu, T.; Walzog, B. Midkine in inflammation. Sci. World J. 2011, 11, 2491–2505. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ikematsu, S.; Moritoyo, T.; Goto, M.; Tsutsui, J.; Sakuma, S.; Osame, M.; Muramatsu, T. Intraventricular administration of the neurotrophic factor midkine ameliorates hippocampal delayed neuronal death following transient forebrain ischemia in gerbils. Brain Res. 2001, 894, 46–55. [Google Scholar] [CrossRef]
- Sato, W.; Kadomatsu, K.; Yuzawa, Y.; Muramatsu, H.; Hotta, N.; Matsuo, S.; Muramatsu, T. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J. Immunol. 2001, 167, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, T.; Shishido, T.; Suzuki, S.; Katoh, S.; Sasaki, T.; Ishino, M.; Nitobe, J.; Miyamoto, T.; Miyashita, T.; Watanabe, T.; et al. Serum midkine as a predictor of cardiac events in patients with chronic heart failure. J. Card. Fail. 2010, 16, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Przybylowski, P.; Malyszko, J.; Malyszko, J.S. Serum midkine is related to NYHA class and cystatin C in heart transplant recipients. Transplant. Proc. 2010, 42, 3704–3707. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Kadomatsu, K. Midkine in the pathology of cancer, neural disease, and inflammation. Pathol. Int. 2012, 62, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Meng, K.; Rodríguez-Peña, A.; Dimitrov, T.; Chen, W.; Yamin, M.; Masaharu Noda, M.; Deuel, T. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc. Natl. Acad. Sci. USA 2000, 97, 2603–2608. [Google Scholar] [CrossRef] [PubMed]
- Liedert, A.; Mattausch, L.; Röntgen, V.; Blakytny, R.; Vogele, D.; Pahl, M.; Bindl, R.; Neunaber, C.; Schinke, T.; Harroch, S.; et al. Midkine-deficiency increases the anabolic response of cortical bone to mechanical loading. Bone 2011, 48, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Ichihara-Tanaka, K.; Kimura, T.; Kadomatsu, K.; Muramatsu, T.; Noda, M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J. Biol. Chem. 1999, 274, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Zou, P.; Suzuki, H.; Oda, Y.; Chen, G-Y.; Sakaguchi, N.; Sakuma, S.; Maeda, N.; Noda, M.; Takada, Y.; et al. α4β1-and α6β1-integrins are functional receptors for midkine, a heparin-binding growth factor. J. Cell Sci. 2004, 117, 5405–5415. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.E.; Wu, Y.; Huang, J.; Zhang, L.; Pratt, R.E.; Dzau, V.J. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol. Biol. Cell 2007, 18, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Matsushita, N. Mechanical stretch and endothelial to mesenchymal transition—Importance of integrin beta1. Circ. J. 2015, 79, 53–54. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Hu, Q.; Xie, Y.; Su, S.; Qiu, Q.; Yuan, W.; Yang, Y.; Song, E.; Chen, Y.; Wang, J. Dyssynchronous pacing triggers endothelial-mesenchymal transition through heterogeneity of mechanical stretch in a canine model. Circ. J. 2015, 79, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Lillis, A.P.; Mikhailenko, I.; Strickland, D.K. Beyond endocytosis: LRP function in cell migration, proliferation and vascular permeability. J. Thromb. Haemost. 2005, 3, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Zou, K.; Sakaguchi, N.; Ikematsu, S.; Sakuma, S.; Muramatsu, T. LDL receptor-related protein as a component of the midkine receptor. Biochem. Biophys. Res. Commun. 2000, 270, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Suh, H.N.; Lee, Y.J.; Seo, B.N.; Ha, J.W.; Han, H.J. Midkine prevented hypoxic injury of mouse embryonic stem cells through activation of Akt and HIF-1alpha via low-density lipoprotein receptor-related protein-1. J. Cell Physiol. 2012, 227, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Kim, D.-H.; Magoori, K.; Fujino, T.; Yamamoto, T.T. A novel low-density lipoprotein receptor-related protein with type II membrane protein-like structure is abundant in heart. J. Biochem. 1998, 124, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Gálvez-Montón, C.; Gonzalo-Calvo de, D.; Valeno, A.G.; Gasterlurruti, P.; Revuelata-López, E.; Prat-Vidal, C.; Soler-Botija, C.; Lluciá-Valldeperas, A.; Perea-Gil, I.; et al. Extracellular vesicles do not contribute to higher circulating levels of soluble LRP1 in idiopathic dilated cardiomyopathy. J. Cell Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hoque, M.O.; Wu, F.; Trink, B.; Sidransky, D.; Ratovitski, E.A. Midkine induces epithelial-mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle 2008, 7, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Kishida, S.; Mu, P.; Miyakawa, S.; Fujiwara, M.; Abe, T.; Sakamoto, K.; Onishi, A.; Nakamura, Y.; Kadomatsu, K. Midkine promotes neuroblastoma through Notch2 signaling. Cancer Res. 2013, 73, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.; Kesavan, A. Novel Heterozygous Mutations in JAG1 and NOTCH2 Genes in a Neonatal Patient with Alagille Syndrome. Case Rep. Pediatr. 2017, 1368189. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Song, G.; Liu, M.; Chen, B.; Chen, Y.; Shen, Y.; Zhu, J.; Zhou, X. MicroRNA-375 overexpression influences P19 cell proliferation, apoptosis and differentiation through the Notch signaling pathway. Int. J. Mol. Med. 2016, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Noseda, M.; Higginson, M.; Ly, M.; Patenaude, A.; Fuller, M.; Kyle, A.H.; Minchinton, A.I.; Puri, M.C.; Dumont, D.J.; et al. Differentiation of vascular smooth muscle cells from local precursors during embryonic and adult arteriogenesis requires Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 6993–6998. [Google Scholar] [CrossRef] [PubMed]
- Gridley, T. Notch signaling in the vasculature. Curr. Top. Dev. Biol. 2010, 92, 277–309. [Google Scholar] [PubMed]
- Campa, V.M.; Gutiérrez-Lanza, R.; Cerignoli, F.; Díaz-Trelles, R.; Nelson, B.; Tsuji, T.; Barcova, M.; Jiang, W.; Mercola, M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J. Cell Biol. 2008, 183, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Muramatsu, T.; Hirai, M.; Inui, T.; Kimura, T.; Saito, H.; McCormick, L.M.; Bu, G.; Kadomatsu, K. Nuclear targeting by the growth factor midkine. Mol. Cell Biol. 2002, 22, 6788–6796. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.C.; Shao, J.-Z.; Min, L.-S.; Xiao, Y.-T.; Xiang, L.-X.; Ma, Z.-H. Midkine accumulated in nucleolus of HepG2 cells involved in rRNA transcription. World J. Gastroenterol. 2008, 14, 6249–6253. [Google Scholar] [CrossRef] [PubMed]
- Tatman, P.D.; Woulfe, K.C.; Karimpour-Fard, A.; Jeffrey, D.A.; Jaggers, J.; Cleveland, J.C.; Nunley, K.; Taylor, M.R.G.; Miyamoto, S.D.; Stauffer, B.L.; et al. Pediatric dilated cardiomyopathy hearts display a unique gene expression profile. JCI Insight 2017, 2, e94249. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Kadomatsu, K.; Yuasa, S.; Muramatsu, H.; Mamiya, T.; Nabeshima, T.; Fan, Q-W.; Ishiguri, K.; Igakura, T.; Matsubara, S.; et al. Disruption of the midkine gene (Mdk) resulted in altered expression of a calcium binding protein in the hippocampus of infant mice and their abnormal behaviour. Genes Cells 1998, 3, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Horiba, M.; Kadomatsu, K.; Yasui, K.; Lee, J.-K.; Takenaka, H.; Sumida, A.; Kamiya, K.; Chen, S.; Sakuma, S.; Muramatsu, T.; et al. Midkine plays a protective role against cardiac ischemia/reperfusion injury through a reduction of apoptotic reaction. Circulation 2006, 114, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Netsu, S.; Shishido, T.; Kitahara, T.; Honda, Y.; Funayama, A.; Narumi, T.; Kadowaki, S.; Takahashi, H.; Miyamoto, T.; Arimoto, T. Midkine exacerbates pressure overload-induced cardiac remodeling. Biochem. Biophys. Res. Commun. 2014, 443, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Zou, P.; Kurosawa, N.; Ichihara-Tanaka, K.; Maruyama, K.; Inoh, K.; Sakai, T.; Chen, L.; Sato, M.; Maramatsu, T. Female infertility in mice deficient in midkine and pleiotrophin, which form a distinct family of growth factors. Genes Cells 2006, 11, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Horiba, M.; Takenaka, H.; Sumida, A.; Opthof, T.; Ishiguro, Y.S.; Kadomatsu, K.; Murohara, T.; Kodama, I. A single intracoronary injection of midkine reduces ischemia/reperfusion injury in Swine hearts: A novel therapeutic approach for acute coronary syndrome. Front. Physiol. 2011, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S.; Matsumoto, K.; Sawa, Y.; Matsuda, H.; Nakamura, T. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J. Clin. Investig. 2000, 106, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Parsa, C.J.; Matsumoto, A.; Kim, J.; Riel, R.U.; Pascal, L.S.; Walton, G.B.; Thompson, R.B.; Petrofski, J.A.; Annex, B.H.; Stamler, J.S.; et al. A novel protective effect of erythropoietin in the infarcted heart. J. Clin. Investig. 2003, 112, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Fukui, S.; Kitagawa-Sakakida, S.; Kawamata, S.; Matsumiya, G.; Kawaguchi, N.; Matsuura, N.; Sawa, Y. Therapeutic effect of midkine on cardiac remodeling in infarcted rat hearts. Ann. Thorac. Surg. 2008, 85, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Sumida, A.; Horiba, M.; Ishiguro, H.; Takenaka, H.; Ueda, N.; Ooboshi, H.; Opthof, T.; Kadomatsu, K.; Kodama, I. Midkine gene transfer after myocardial infarction in rats prevents remodelling and ameliorates cardiac dysfunction. Cardiovasc. Res. 2010, 86, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, H.; Horiba, M.; Ishiguro, H.; Sumida, A.; Hojo, M.; Usui, A.; Akita, T.; Sakuma, S.; Ueda, Y.; Kodama, I.; et al. Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H462–H469. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Hojo, M.; Kamiya, K.; Kadomatsu, K.; Murohara, T.; Kodama, I.; Horiba, M. Exogenous midkine administration prevents cardiac remodeling in pacing-induced congestive heart failure of rabbits. Heart Vessels 2016, 31, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Shishido, T.; Takahashi, T.; Watanabe, T.; Netsu, S.; Kinoshita, D.; Narumi, T.; Kadowaki, S.; Nishiyama, S.; Takahashi, H.; et al. Midkine Deteriorates Cardiac Remodeling via Epidermal Growth Factor Receptor Signaling in Chronic Kidney Disease. Hypertension 2016, 67, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Hobo, A.; Yuzawa, Y.; Kosugi, T.; Kato, N.; Asai, N.; Sato, W.; Maruyama, S.; Ito, Y.; Kobori, H.; Ikematsu, S.; et al. The growth factor midkine regulates the renin-angiotensin system in mice. J. Clin. Investig. 2009, 119, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra, L.; Herradon, G.; Nguyen, T.; Silos-Santiago, I.; Deuel, T.F. Midkine is a potent regulator of the catecholamine biosynthesis pathway in mouse aorta. Life Sci. 2006, 79, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Laake, L.W.V.; Davidson, S.M.; Engel, F.B.; Hausenloy, D.J.; Lecour, S.; Leor, J.; Perrino, C.; Schulz, R.; Ytrehus, K.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.W.; Yu, S.-L.; Chen, W.-J.; Yang, P.-C.; Chien, C.-T.; Chou, H.-Y.; Li, H.-N.; Peck, K.; Huang, C.-H.; Lin, F.-Y.; et al. Dynamic changes of gene expression profiles during postnatal development of the heart in mice. Heart 2004, 90, 927–934. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease Model | Study | Animal Model | Changes in Circulating MDK | Changes in Myocardial MDK | Outcome |
|---|---|---|---|---|---|
| Ischemia/Reperfusion | Horiba et al. (2006) | ♂ WT vs. MDK KO mice (12 weeks old) —IM injection of 0.008 mg/kg MDK | Not studied | WT: MDK protein peak 24 h post I/R | KO: Higher mortality, worse pathology KO + MDK: decreased infarct size |
| Ishiguro et al. (2011) | ♂ pigs (adult) —intracoronary injection of 0.005 mg/kg MDK | Not studied; extracellular MDK in LV in peri-infarct area | Not studied | MDK tx decreased infarct size and apoptosis | |
| Myocardial Infarction | Fukui et al. (2007) | ♂ Wistar Rats (8 weeks old) —3 doses (0.021 mg/kg; 0.12 mg/kg; 0.53 mg/kg) MDK injected in border zone | Not studied | MDK gene expression peaks 7 days post MI and MDK protein is present in border zone | MDK tx improved function, increased non-infarcted area, and increased angiogenesis in a dose-dependent manner |
| Sumida et al. (2010) | ♂ Wistar Rats (8–10 weeks old) —30 min after ligation inject AdMDK IM (5–6 sites; 1 × 109 PFU) | Not studied | Sham + AdMDK: high MDK protein until 8 weeks | AdMDK tx lead to improved function at 4 weeks post MI; at 6 weeks post MI: decreased collagen in non-infarct, increased angiogenesis | |
| Takenaka et al. (2009) | ♂ WT vs. MDK KO mice (12 weeks old) —MDK administered in osmotic pumps at time of MI (4.25 mg/kg/week) | Not studied | WT: MDK expression is 7-fold higher than sham at 14 days post MI (protein MDK also increases by day 14) KO + MDK: increased MDK in peri-infarct cardiomyocytes | KO: Higher mortality KO + MDK: increased survival WT + MDK: increased survival, decreased BNP, reduced decline in function; less fibrosis, increased angiogenesis | |
| Thoracic Aortic Constriction | Netsu et al. (2014) | ♂ WT vs. MDK TG mice (8–10 weeks old) | Not studied | WT: MDK mRNA peaks 14 days post TAC; lung and kidney mRNA and protein elevated at 14–28 days | MDK TG have higher mortality decreased function, higher HW/BW, more fibrosis |
| Rapid pacing | Harada et al. (2014) | ♂ Rabbits (2 kg) —MDK delivered by osmotic pump (1 mg/kg/week) | Not studied | Not studied | MDK tx decreased mortality; increased function, decreased apoptosis |
| Kidney dysfunction | Honda et al. (2016) | ♂ MDK KO mice subtotal nephrectomy | Not studied | Not studied | MDK KO decreased cardiac hypertrophy |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woulfe, K.C.; Sucharov, C.C. Midkine’s Role in Cardiac Pathology. J. Cardiovasc. Dev. Dis. 2017, 4, 13. https://doi.org/10.3390/jcdd4030013
Woulfe KC, Sucharov CC. Midkine’s Role in Cardiac Pathology. Journal of Cardiovascular Development and Disease. 2017; 4(3):13. https://doi.org/10.3390/jcdd4030013
Chicago/Turabian StyleWoulfe, Kathleen C., and Carmen C. Sucharov. 2017. "Midkine’s Role in Cardiac Pathology" Journal of Cardiovascular Development and Disease 4, no. 3: 13. https://doi.org/10.3390/jcdd4030013
APA StyleWoulfe, K. C., & Sucharov, C. C. (2017). Midkine’s Role in Cardiac Pathology. Journal of Cardiovascular Development and Disease, 4(3), 13. https://doi.org/10.3390/jcdd4030013