
The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming


Abstract
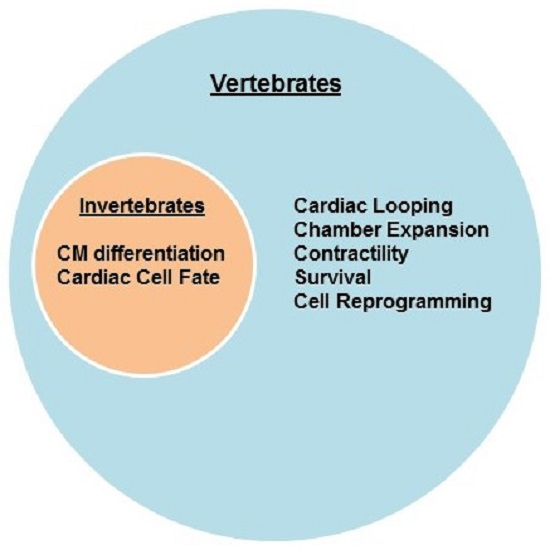
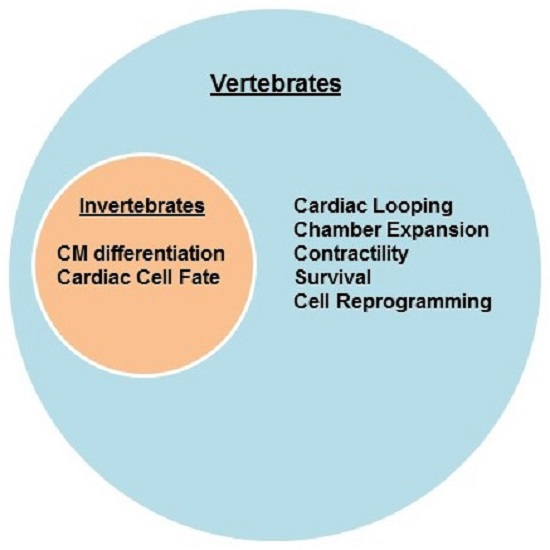
:
1. Introduction
2. Cardiac Development and Differentiation—Model Organisms
2.1. Invertebrates
2.1.1. Flies (D. melanogaster)
Function
2.1.2. Nematodes (C. elegans)
2.1.3. Ascidians (C. intestinalis)
2.2. Vertebrates
2.2.1. Zebrafish (D. rerio)
Function
2.2.2. Frogs (X. laevis)
Function
2.2.3. Avian (Gallus gallus)
2.3. Rodents—Mice (M. musculus), Rat (R. norvegicus)
2.3.1. MEF2A
2.3.2. MEF2C
2.3.3. MEF2D
2.3.4. Additional Functional Studies Relating to MEF2 Misexpression and Activity
3. Functional Genomic Analysis of MEF2 in Striated Muscle
4. Direct Cardiomyocyte Reprogramming/Transdifferentiation
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McCulley, D.J.; Black, B.L. Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar]
- Clark, K.L.; Yutzey, K.E.; Benson, D.W. Transcription factors and congenital heart defects. Annu. Rev. Physiol. 2006, 68, 97–121. [Google Scholar] [PubMed]
- Oka, T.; Xu, J.; Molkentin, J.D. Re-employment of developmental transcription factors in adult heart disease. Semin. Cell Dev. Biol. 2007, 18, 117–131. [Google Scholar] [PubMed]
- Black, B.L.; Olson, E.N. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 1998, 14, 167–196. [Google Scholar] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Development 2007, 134, 4131–4140. [Google Scholar] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. MEF2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 2002, 27, 40–47. [Google Scholar]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Gulick, T. Phosphorylation and alternative pre-mRNA splicing converge to regulate myocyte enhancer factor 2C activity. Mol. Cell Biol. 2004, 24, 8264–8275. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, A.L.; Cripps, R.M. Cardiac gene regulatory networks in Drosophila. Biochim. Biophys. Acta 2009, 1789, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Vogler, G.; Bodmer, R. Cellular Mechanisms of Drosophila Heart Morphogenesis. J. Cardiovasc. Dev. Dis. 2015, 2, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Lilly, B.; Galewsky, S.; Firulli, A.B.; Schulz, R.A.; Olson, E.N. D-MEF2: A MADS box transcription factor expressed in differentiating mesoderm and muscle cell lineages during Drosophila embryogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 5662–5666. [Google Scholar] [CrossRef] [PubMed]
- Lilly, B.; Zhao, B.; Ranganayakulu, G.; Paterson, B.M.; Schulz, R.A.; Olson, E.N. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science 1995, 267, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Bour, B.A.; O’Brien, M.A.; Lockwood, W.L.; Goldstein, E.S.; Bodmer, R.; Taghert, P.H.; Abmayr, S.M.; Nguyen, H.T. Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes Dev. 1995, 9, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Lovato, T.L.; Sensibaugh, C.A.; Swingle, K.L.; Martinez, M.M.; Cripps, R.M. The Drosophila Transcription Factors Tinman and Pannier Activate and Collaborate with Myocyte Enhancer Factor-2 to Promote Heart Cell Fate. PLoS ONE 2015, 10, e0132965. [Google Scholar]
- Dichoso, D.; Brodigan, T.; Chwoe, K.Y.; Lee, J.S.; Llacer, R.; Park, M.; Corsi, A.K.; Kostas, S.A.; Fire, A.; Ahnn, J.; et al. The MADS-Box factor CeMEF2 is not essential for Caenorhabditis elegans myogenesis and development. Dev. Biol. 2000, 223, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Cota, C.D.; Segade, F.; Davidson, B. Heart genetics in a small package, exploiting the condensed genome of Ciona intestinalis. Brief. Funct. Genom. 2014, 13, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.S.; Hino, K.; Yagi, K.; Satoh, N.; Satou, Y. Gene expression profiles of transcription factors and signaling molecules in the ascidian embryo: Towards a comprehensive understanding of gene networks. Development 2004, 131, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.; Christoffels, V.M. Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 2003, 83, 1223–1267. [Google Scholar] [CrossRef] [PubMed]
- Ticho, B.S.; Stainier, D.Y.; Fishman, M.C.; Breitbart, R.E. Three zebrafish MEF2 genes delineate somitic and cardiac muscle development in wild-type and mutant embryos. Mech. Dev. 1996, 59, 205–218. [Google Scholar] [CrossRef]
- Lazic, S.; Scott, I.C. Mef2cb regulates late myocardial cell addition from a second heart field-like population of progenitors in zebrafish. Dev. Biol. 2011, 354, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hinits, Y.; Hughes, S.M. Mef2s are required for thick filament formation in nascent muscle fibres. Development 2007, 134, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Hinits, Y.; Pan, L.; Walker, C.; Dowd, J.; Moens, C.B.; Hughes, S.M. Zebrafish Mef2ca and Mef2cb are essential for both first and second heart field cardiomyocyte differentiation. Dev. Biol. 2012, 369, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Qian, L.X.; Yu, Z.; Jiang, Q.; Dong, Y.X.; Liu, X.F.; Yang, X.Y.; Zhong, T.P.; Song, H.Y. Requirements of myocyte-specific enhancer factor 2A in zebrafish cardiac contractility. FEBS Lett. 2005, 579, 4843–4850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Qian, L.X.; Liu, D.; Yao, L.L.; Jiang, Q.; Yu, Z.; Gui, Y.H.; Zhong, T.P.; Song, H.Y. Bone morphogenetic protein-2 acts upstream of myocyte-specific enhancer factor 2a to control embryonic cardiac contractility. Cardiovasc. Res. 2007, 74, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Abboud, S.L.; Mahimainathan, L.; Chandrasekar, B.; Choudhury, G.G. Phosphatidylinositol 3-kinase regulates bone morphogenetic protein-2 (BMP-2)-induced myocyte enhancer factor 2A-dependent transcription of BMP-2 gene in cardiomyocyte precursor cells. J. Biol. Chem. 2003, 278, 21998–22005. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.E.; Kotecha, S.; Towers, N.; Mohun, T.J. Muscle-specific expression of SRF-related genes in the early embryo of Xenopus laevis. EMBO J. 1992, 11, 4981–4991. [Google Scholar] [PubMed]
- Chambers, A.E.; Logan, M.; Kotecha, S.; Towers, N.; Sparrow, D.; Mohun, T.J. The RSRF/MEF2 protein SL1 regulates cardiac muscle-specific transcription of a myosin light-chain gene in Xenopus embryos. Genes Dev. 1994, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Gessert, S.; Kühl, M. Comparative gene expression analysis and fate mapping studies suggest an early segregation of cardiogenic lineages in Xenopus laevis. Dev. Biol. 2009, 334, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kühl, S.J.; Pfister, A.S.; Cizelsky, W.; Denk, S.; Beer-Molz, L.; Kühl, M. Comparative analysis reveals distinct and overlapping functions of Mef2c and Mef2d during cardiogenesis in Xenopus laevis. PLoS ONE 2014, 9, e87294. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Izumo, S. Cardiac myogenesis: Overexpression of XCsx2 or XMEF2A in whole Zenopus embryos induces the precocious expression of XMHCalpha gene. Roux’s Arch. Dev. Biol. 1995, 205, 198–202. [Google Scholar] [CrossRef]
- Berti, F.; Nogueira, J.M.; Wöhrle, S.; Sobreira, D.R.; Hawrot, K.; Dietrich, S. Time course and side-by-side analysis of mesodermal, pre-myogenic, myogenic and differentiated cell markers in the chicken model for skeletal muscle formation. J. Anat. 2015, 227, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A.; Arnold, H.H. The MADS domain containing transcription factor cMef2a is expressed in heart and skeletal muscle during embryonic chick development. Dev. Genes Evol. 1999, 209, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi-Suzuki, K.; Pauliks, L.B.; Eltsefon, Y.; Mikawa, T. Purkinje fibers of the avian heart express a myogenic transcription factor program distinct from cardiac and skeletal muscle. Dev. Biol. 2001, 234, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.G.; Lyons, G.E.; Martin, J.F.; Olson, E.N. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development 1994, 120, 1251–1263. [Google Scholar] [PubMed]
- Molkentin, J.D.; Firulli, A.B.; Black, B.L.; Martin, J.F.; Hustad, C.M.; Copeland, N.; Jenkins, N.; Lyons, G.; Olson, E.N. MEF2B is a potent transactivator expressed in early myogenic lineages. Mol. Cell Biol. 1996, 16, 3814–3824. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.H.; Clemente, C.F.; Cardoso, A.C.; Theizen, T.H.; Rocco, S.A.; Judice, C.C.; Guido, M.C.; Pascoal, V.D.; Lopes-Cendes, I.; Souza, J.R.; et al. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS ONE 2009, 4, e8472. [Google Scholar] [CrossRef] [PubMed]
- Naya, F.J.; Wu, C.; Richardson, J.A.; Overbeek, P.; Olson, E.N. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development 1999, 126, 2045–2052. [Google Scholar] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Olson, E.N. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 2001, 11, 497–504. [Google Scholar] [CrossRef]
- Naya, F.J.; Black, B.L.; Wu, H.; Bassel-Duby, R.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Durham, J.T.; Brand, O.M.; Arnold, M.; Reynolds, J.G.; Muthukumar, L.; Weiler, H.; Richardson, J.A.; Naya, F.J. Myospryn is a direct transcriptional target for MEF2A that encodes a striated muscle, alpha-actinin-interacting, costamere-localized protein. J. Biol. Chem. 2006, 281, 6841–6849. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.T.; Brand, O.M.; Mathew, M.; Ignatiou, C.; Ewen, E.P.; McCalmon, S.A.; Naya, F.J. Myomaxin is a novel transcriptional target of MEF2A that encodes a Xin-related alpha-actinin-interacting protein. J. Biol. Chem. 2006, 281, 39370–39379. [Google Scholar] [CrossRef] [PubMed]
- Ewen, E.P.; Snyder, C.M.; Wilson, M.; Desjardins, D.; Naya, F.J. Mef2A coordinately regulates a costamere gene program in cardiac muscle. J. Biol. Chem. 2011, 286, 29644–29653. [Google Scholar] [CrossRef] [PubMed]
- Kachigian, L.M. Early growth response-1 in cardiovascular pathobiology. Circ. Res. 2006, 98, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Desjardins, C.A.; Cooper, O.; Kontor, A.; Nocco, S.E.; Naya, F.J. EGR1 Functions as a Potent Repressor of MEF2 Transcriptional Activity. PLoS ONE 2015, 10, e0127641. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wu, X.; Druso, J.E.; Wei, H.; Park, A.Y.; Kraus, M.S.; Alcaraz, A.; Chen, J.; Chien, S.; Cerione, R.A.; et al. Cardiac developmental defects and eccentric right ventricular hypertrophy incardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6638–6643. [Google Scholar] [CrossRef] [PubMed]
- Nadruz, W., Jr.; Corat, M.A.; Marin, T.M.; Guimarães Pereira, G.A.; Franchini, K.G. Focal adhesion kinase mediates MEF2 and c-Jun activation by stretch: Role in the activation of the cardiac hypertrophic genetic program. Cardiovasc. Res. 2005, 68, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Pereira, A.H.; Ambrosio, A.L.; Consonni, S.R.; Rocha de Oliveira, R.; Bajgelman, M.C.; Dias, S.M.; Franchini, K.G. FAK Forms a Complex with MEF2 to Couple Biomechanical Signaling to Transcription in Cardiomyocytes. Structure 2016, 24, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Lu, J.; Yanagisawa, H.; Webb, R.; Lyons, G.E.; Richardson, J.A.; Olson, E.N. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development 1998, 125, 4565–4574. [Google Scholar] [PubMed]
- Bi, W.; Drake, C.J.; Schwarz, J.J. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev. Biol. 1999, 211, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Vong, L.H.; Ragusa, M.J.; Schwarz, J.J. Generation of conditional Mef2cloxP/loxP mice for temporal- and tissue-specific analyses. Genesis 2005, 43, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Vincentz, J.W.; Barnes, R.M.; Firulli, B.A.; Conway, S.J.; Firulli, A.B. Cooperative interaction of Nkx2.5 and Mef2c transcription factors during heart development. Dev. Dyn. 2008, 237, 3809–3819. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.M.; Harris, I.S.; Jaehnig, E.J.; Sauls, K.; Sinha, T.; Rojas, A.; Schachterle, W.; McCulley, D.J.; Norris, R.A.; Black, B.L. MEF2C regulates outflow tract alignment and transcriptional control of Tdgf1. Development 2016, 143, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L. Transcriptional pathways in second heart field development. Semin. Cell Dev. Biol. 2007, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z.; Valdez, M.R.; McAnally, J.; Richardson, J.; Olson, E.N. The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development 2001, 128, 4623–4633. [Google Scholar] [PubMed]
- Dodou, E.; Verzi, M.P.; Anderson, J.P.; Xu, S.M.; Black, B.L. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 2004, 131, 3931–3942. [Google Scholar] [CrossRef] [PubMed]
- De Val, S.; Chi, N.C.; Meadows, S.M.; Minovitsky, S.; Anderson, J.P.; Harris, I.S.; Ehlers, M.L.; Agarwal, P.; Visel, A.; Xu, S.M.; et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell 2008, 135, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Von Both, I.; Silvestri, C.; Erdemir, T.; Lickert, H.; Walls, J.R.; Henkelman, R.M.; Rossant, J.; Harvey, R.P.; Attisano, L.; Wrana, J.L. Foxh1 is essential for development of the anterior heart field. Dev. Cell. 2004, 7, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, B.; Yu, G.; Li, S.; Zhu, B.; Gulick, T. Myocyte enhancer factor 2A is transcriptionally autoregulated. J. Biol. Chem. 2008, 283, 10318–10329. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Phan, D.; van Rooij, E.; Wang, D.Z.; McAnally, J.; Qi, X.; Richardson, J.A.; Hill, J.A.; Bassel-Duby, R.; Olson, E.N. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J. Clin. Investig. 2008, 118, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Estrella, N.L.; Clark, A.L.; Desjardins, C.A.; Nocco, S.E.; Naya, F.J. MEF2D Deficiency in Neonatal Cardiomyocytes Triggers Cell Cycle Re-entry and Programmed Cell Death in vitro. J. Biol. Chem. 2015, 290, 24367–24380. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Faralli, H.; Yao, Z.; Rakopoulos, P.; Palii, C.; Cao, Y.; Singh, K.; Liu, Q.C.; Chu, A.; Aziz, A.; et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 2013, 27, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Perry, R.L.; Nowacki, N.B.; Gordon, J.W.; Salma, J.; Zhao, J.; Aziz, A.; Chan, J.; Siu, K.W.; McDermott, J.C. Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol. Cell. Biol. 2008, 28, 2952–2970. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ramachandran, B.; Gulick, T. Alternative pre-mRNA splicing governs expression of a conserved acidic transactivation domain in myocyte enhancer factor 2 factors of striated muscle and brain. J. Biol. Chem. 2005, 280, 28749–28760. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Huang, J.; Hamamori, Y.; Kedes, L. Molecular mechanisms of myogenic coactivation by p300: Direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell. Biol. 1997, 17, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- Miska, E.A.; Karlsson, C.; Langley, E.; Nielsen, S.J.; Pines, J.; Kouzarides, T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999, 18, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, D.B.; Miska, E.A.; Langley, E.; Reynaud-Deonauth, S.; Kotecha, S.; Towers, N.; Spohr, G.; Kouzarides, T.; Mohun, T.J. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999, 18, 5085–5098. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Olson, E.N. Toward transcriptional therapies for the failing heart: Chemical screens to modulate genes. J. Clin. Investig. 2005, 115, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Backs, J.; Olson, E.N. Control of cardiac growth by histone acetylation/deacetylation. Circ. Res. 2006, 98, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Parra, M.; Verdin, E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 2010, 10, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, L.H.; Worst, B.C.; Stanmore, D.A.; Backs, J. Histone deacetylase signaling in cardioprotection. Cell. Mol. Life Sci. 2014, 71, 1673–1690. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell. 2008, 32, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Karamboulas, C.; Swedani, A.; Ward, C.; Al-Madhoun, A.S.; Wilton, S.; Boisvenue, S.; Ridgeway, A.G.; Skerjanc, I.S. HDAC activity regulates entry of mesoderm cells into the cardiac muscle lineage. J. Cell Sci. 2006, 119, 4305–4314. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Kim, J.; Anderson, J.P.; Wu, J.; Gleim, S.R.; Kundu, R.K.; McLean, D.L.; Kim, J.D.; Park, H.; Jin, S.W.; et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ. Res. 2013, 113, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Karamboulas, C.; Dakubo, G.D.; Liu, J.; DeRepentigny, Y.; Yutzey, K.; Wallace, V.A.; Kothary, R.; Skerjanc, I.S. Disruption of MEF2 activity in cardiomyoblasts inhibits cardiomyogenesis. J. Cell. Sci. 2006, 119, 4315–4321. [Google Scholar] [CrossRef] [PubMed]
- Skerjanc, I.S.; Petropoulos, H.; Ridgeway, A.G.; Wilton, S. Myocyte enhancer factor 2C and Nkx2–5 up-regulate each other’s expression and initiate cardiomyogenesis in P19 cells. J. Biol. Chem. 1998, 273, 34904–34910. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, S.M.; Wang, L.; Balazsi, K.; DeRepentigny, Y.; Kothary, R.; Megeney, L.A. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr. Biol. 1999, 9, 1203–1206. [Google Scholar] [CrossRef]
- Van Oort, R.J.; van Rooij, E.; Bourajjaj, M.; Schimmel, J.; Jansen, M.A.; van der Nagel, R.; Doevendans, P.A.; Schneider, M.D.; van Echteld, C.J.; De Windt, L.J. MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation 2006, 114, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gong, N.L.; Bodi, I.; Aronow, B.J.; Backx, P.H.; Molkentin, J.D. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J. Biol. Chem. 2006, 281, 9152–9162. [Google Scholar] [CrossRef] [PubMed]
- Estrella, N.L.; Naya, F.J. Transcriptional networks regulating the costamere, sarcomere, and other cytoskeletal structures in striated muscle. Cell. Mol. Life Sci. 2014, 71, 1641–1656. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, T.; Jensen, L.J.; Jakobsen, J.S.; Karzynski, M.M.; Eichenlaub, M.P.; Bork, P.; Furlong, E.E. A temporal map of transcription factor activity: Mef2 directly regulates target genes at all stages of muscle development. Dev. Cell. 2006, 10, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Junion, G.; Jagla, T.; Duplant, S.; Tapin, R.; Da Ponte, J.P.; Jagla, K. Mapping Dmef2-binding regulatory modules by using a ChIP-enriched in silico targets approach. Proc. Natl. Acad. Sci. USA 2005, 102, 18479–18484. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.; Virtanen, C.; Lu, Z.; Takahashi, M. Identification of MEF2-regulated genes during muscle differentiation. Physiol. Genom. 2004, 20, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Blais, A.; Tsikitis, M.; Acosta-Alvear, D.; Sharan, R.; Kluger, Y.; Dynlacht, B.D. An initial blueprint for myogenic differentiation. Genes Dev. 2005, 19, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Estrella, N.L.; Desjardins, C.A.; Nocco, S.E.; Clark, A.L.; Maksimenko, Y.; Naya, F.J. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J. Biol. Chem. 2015, 290, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, J.; Schueler, M.; Grunert, M.; Fischer, J.J.; Zhang, Q.; Krueger, T.; Lange, M.; Tönjes, M.; Dunkel, I.; Sperling, S.R. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011, 7, e1001313. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Kong, S.W.; Ma, Q.; Pu, W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. USA 2011, 108, 5632–5637. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Aneas, I.; Sakabe, N.; Dirschinger, R.J.; Wang, G.; Smemo, S.; Westlund, J.M.; Cheng, H.; Dalton, N.; Gu, Y.; et al. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J. Clin. Investig. 2011, 121, 4640–4654. [Google Scholar] [CrossRef] [PubMed]
- Wales, S.; Hashemi, S.; Blais, A.; McDermott, J.C. Global MEF2 target gene analysis in cardiac and skeletal muscle reveals novel regulation of DUSP6 by p38MAPK-MEF2 signaling. Nucleic Acids Res. 2014, 42, 11349–11362. [Google Scholar] [CrossRef] [PubMed]
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Viganò, V.; Stirparo, G.G.; Latronico, M.V.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169. [Google Scholar] [CrossRef] [PubMed]
- Czubryt, M.P.; Olson, E.N. Balancing contractility and energy production: The role of myocyte enhancer factor 2 (MEF2) in cardiac hypertrophy. Recent Prog. Horm. Res. 2004, 59, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, L.; Sakabe, N.J.; Blanski, A.A.; Arimura, F.E.; Westlund, J.M.; Nobrega, M.A.; Ovcharenko, I. Genome-wide discovery of human heart enhancers. Genome Res. 2010, 20, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Krane, M.; Deutsch, M.A.; Wang, L.; Rav-Acha, M.; Gregoire, S.; Engels, M.C.; Rajarajan, K.; Karra, R.; Abel, E.D.; et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
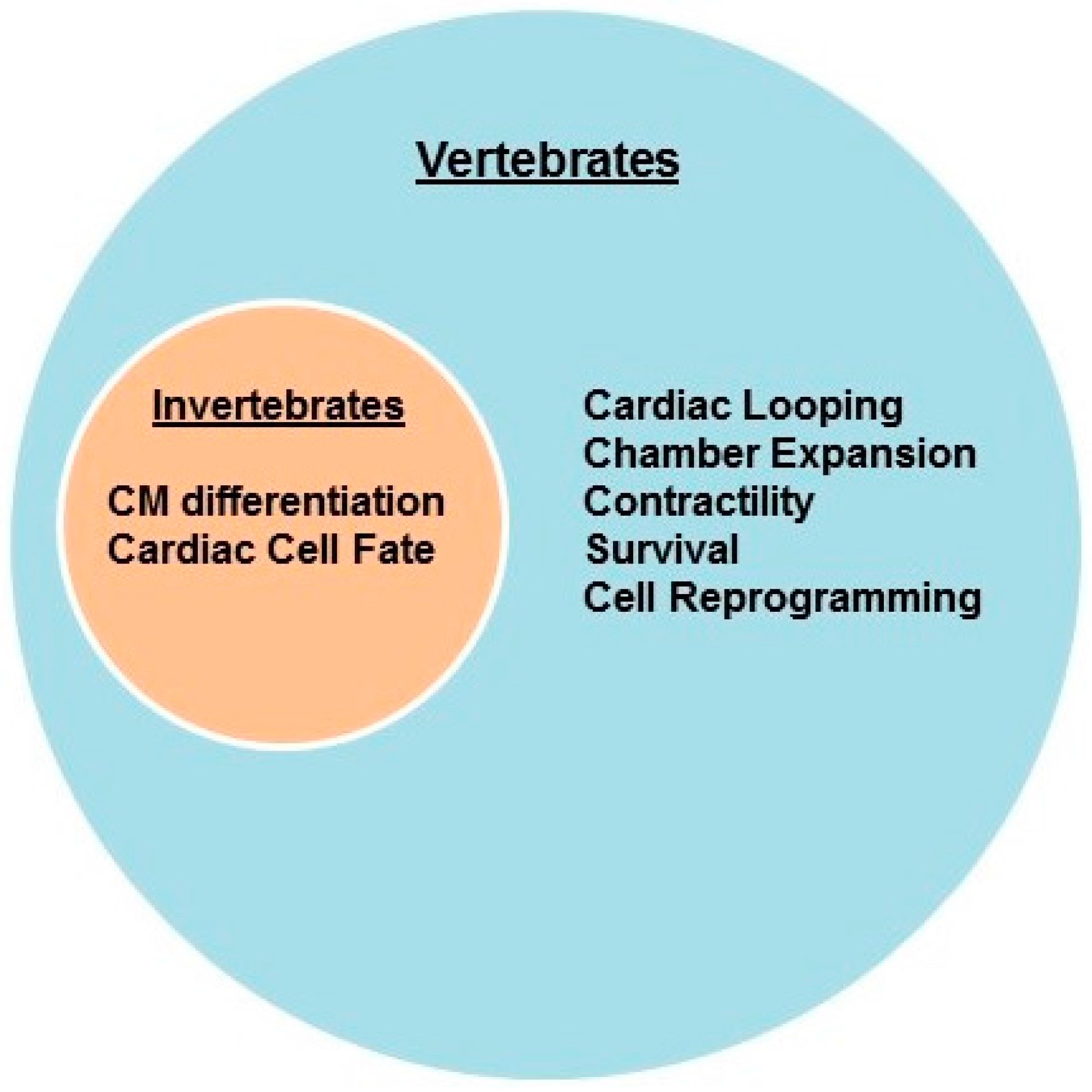

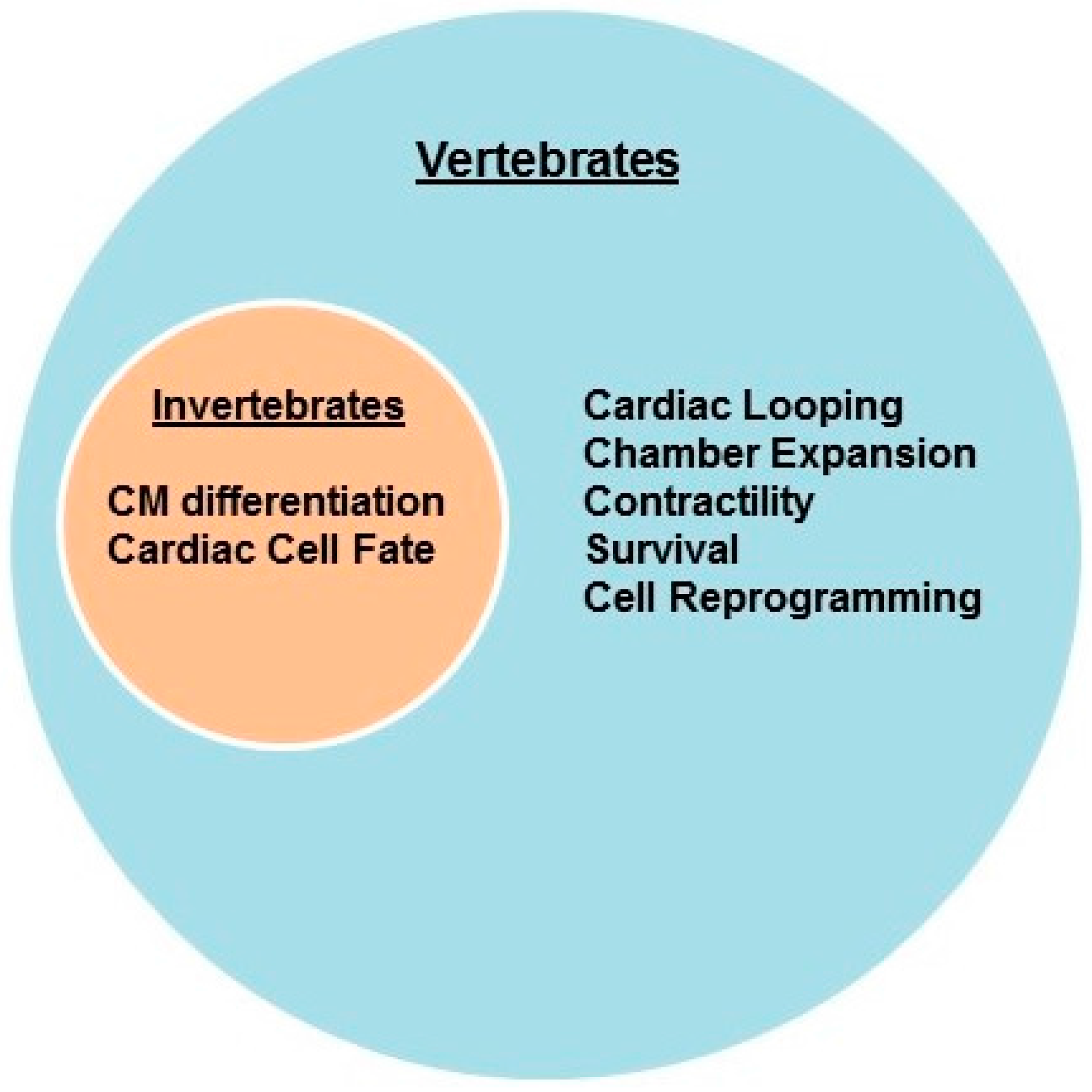
{kind=link}
{kind=link}
| Model | MEF2 Isoform | Genetic Manipulation | Phenotype | Ref |
|---|---|---|---|---|
| D. melanogaster | D-Mef2 | Global Loss-of-Function (LoF) | Cardioblasts are specified, failure to differentiate | [12,13] |
| C. elegans | CeMef2 | Deletion | No observed phenotype | [15] |
| D. rerio | mef2aa | Morpholino knockdown | Normal cardiac morphology | [23] |
| Significant decrease in cardiac contractility | ||||
| Morpholino knockdown (Bmp2) | Significant loss of Mef2a expression | [24] | ||
| Decrease in cardiac contractility rescued by Mef2a overexpression | ||||
| mef2ca | Morpholino knockdown | Delayed cardiomyocyte marker expression | [22] | |
| Cardiac development delayed, normal heart | ||||
| Global LoF | Delayed cardiomyocyte marker expression | [22] | ||
| Cardiac development delayed, normal heart | ||||
| mef2cb | Morpholino knockdown | Secondary heart field defects | [20,22] | |
| Loss of cardiomyocytes in arterial poles | ||||
| Chamber shortening | ||||
| Global LoF | No observed phenotype | [22] | ||
| mef2ca/cb | Global LoF | Pericardial edema | [22] | |
| Impaired cardiomyocyte differentation | ||||
| Impaired heart tube formation | ||||
| All mef2 | Morpholino knockdown | Loss of cardiomyocyte differentiation | [22] | |
| Lack of α-MHC expression | ||||
| X. laevis | mef2a | mRNA microinjection | Precocious expression of α-MHC | [30] |
| Enlarged heart | ||||
| mef2c | Morpholino knockdown | Cardiac looping defect | [29] | |
| Chamber expansion defect | ||||
| Mef2D overexpression rescues phenotype | ||||
| mef2d | Morpholino knockdown | Cardiac looping defect | [29] | |
| Chamber expansion defect | ||||
| Mef2C overexpression fails to rescue phenotype |
| Model | MEF2 Isoform | Genetic Manipulation | Phenotype | Ref |
|---|---|---|---|---|
| M. musculus | Mef2a | Global Loss-of-Function (LoF) | 80% Perinatal lethality | [39,42] |
| Severe myofibrillary defects | ||||
| Dysregulated costamere gene expression | ||||
| 20% Survival to adulthood | [39,42] | |||
| Mitochondrial deficiency | ||||
| Conduction abnormalities | ||||
| Cardiac-specific LoF(FAK) | Signficiant downregulation of Mef2a | [45] | ||
| Embryonic lethality, defective chamber wall maturation, reduced cardiomyocyte proliferation | ||||
| Cardiac overexpression | Dilated cardiomyopathy | [40,82,83] | ||
| Mef2c | Global LoF | Embryonic lethality | [48,49,50] | |
| Defective cardiac looping morphogenesis | ||||
| Vascular malformations | ||||
| CM-specific LoF @~E10.5 | Viable embryo | [51] | ||
| Normal cardiac development | ||||
| Double LoF (Mef2c/Nkx2.5) | Development of a single chamber heart | [52] | ||
| Expression of atrial and secondary heart field markers | ||||
| SHF LoF | Outflow tract defects | [53] | ||
| Overriding aorta and double outlet right ventricle | ||||
| Cardiac overexpression | Dilated cardiomyopathy | [83] | ||
| Mef2d | Global LoF | Viable , normal cardiac structure and function | [62] | |
| Attenuated hypertrophy and fibrosis in response to stress | ||||
| Cardiac overexpression | Atrial enlargement | [62] | ||
| Extensive fibrosis | ||||
| All Mef2 | Dominant Negative | In vitro: impaired cardiomycyte differentiation | [79] | |
| In vivo: failure to form a heart (severe) | ||||
| thin-walled myocardium (mild) | ||||
| Transgenic Dominant Negative | Attenuated myocardial growth | [81] | ||
| No observable phenotype | [82] | |||
| R. norvegicus | Mef2a | shRNA depletion | Costamere dysregulation | [42] |
| Cell Death | ||||
| Mef2d | shRNA depletion | Cell cycle re-entry | [63] | |
| Apoptosis |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desjardins, C.A.; Naya, F.J. The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming. J. Cardiovasc. Dev. Dis. 2016, 3, 26. https://doi.org/10.3390/jcdd3030026
Desjardins CA, Naya FJ. The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming. Journal of Cardiovascular Development and Disease. 2016; 3(3):26. https://doi.org/10.3390/jcdd3030026
Chicago/Turabian StyleDesjardins, Cody A., and Francisco J. Naya. 2016. "The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming" Journal of Cardiovascular Development and Disease 3, no. 3: 26. https://doi.org/10.3390/jcdd3030026
APA StyleDesjardins, C. A., & Naya, F. J. (2016). The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming. Journal of Cardiovascular Development and Disease, 3(3), 26. https://doi.org/10.3390/jcdd3030026