

Doxorubicin-Induced Cardiotoxicity: A Comprehensive Update


 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Doxorubicin: Pharmacokinetics and Pharmacodynamics
3. Doxorubicin in Cardiac Tissue
4. Acute and Chronic Cardiotoxicity
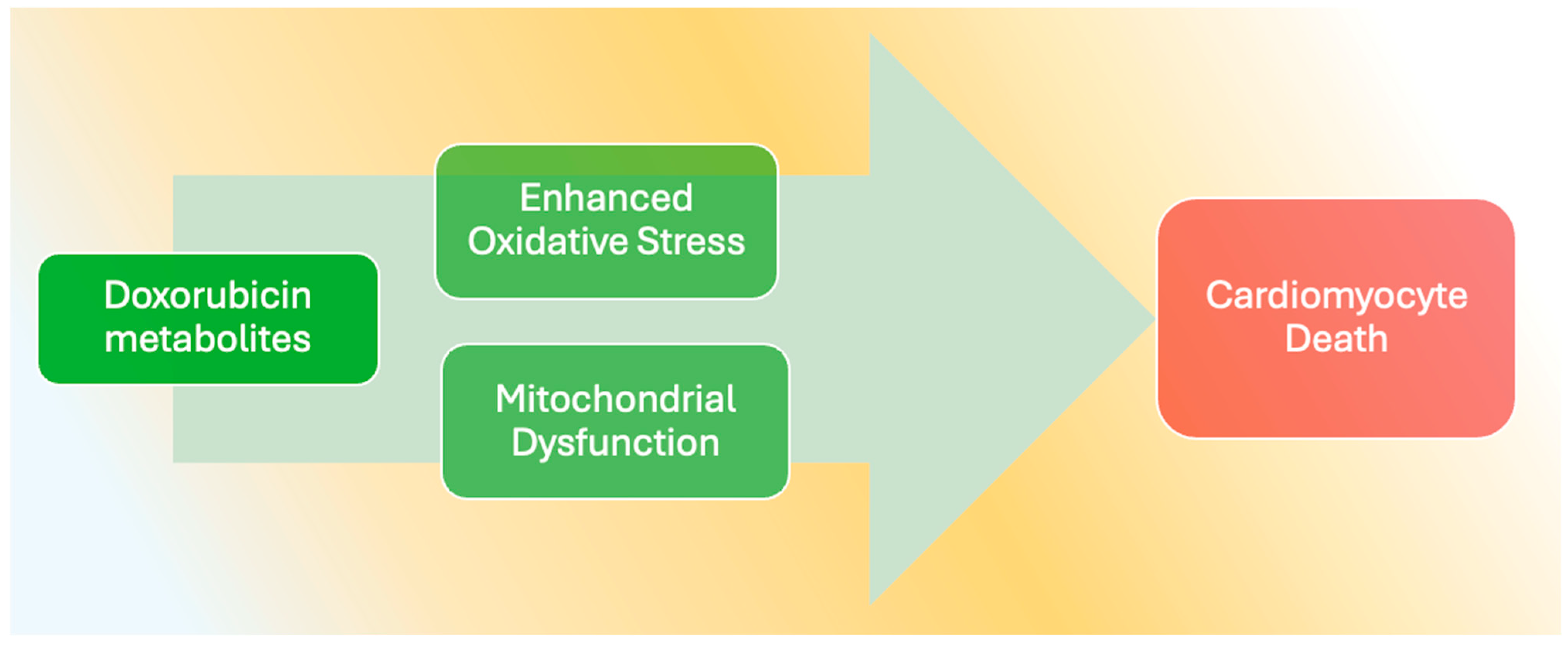
5. Doxorubicin and Cardiomyocyte Death
6. Oxidative Stress
7. Doxorubicin and Calcium in Cardiomyocytes
8. Mitochondrial Dysfunction in Doxorubicin-Treated Hearts
9. Inflammatory Pathways and Immune Modulation
10. Genetic and Epigenetic Susceptibility Factors
11. Pediatric Versus Adult Cardiotoxicity Profiles
12. Structural and Functional Cardiac Changes Detected by Imaging
13. Biomarkers of Cardiotoxicity in Doxorubicin-Treated Patients
14. Endothelial Dysfunction
15. Preclinical Models
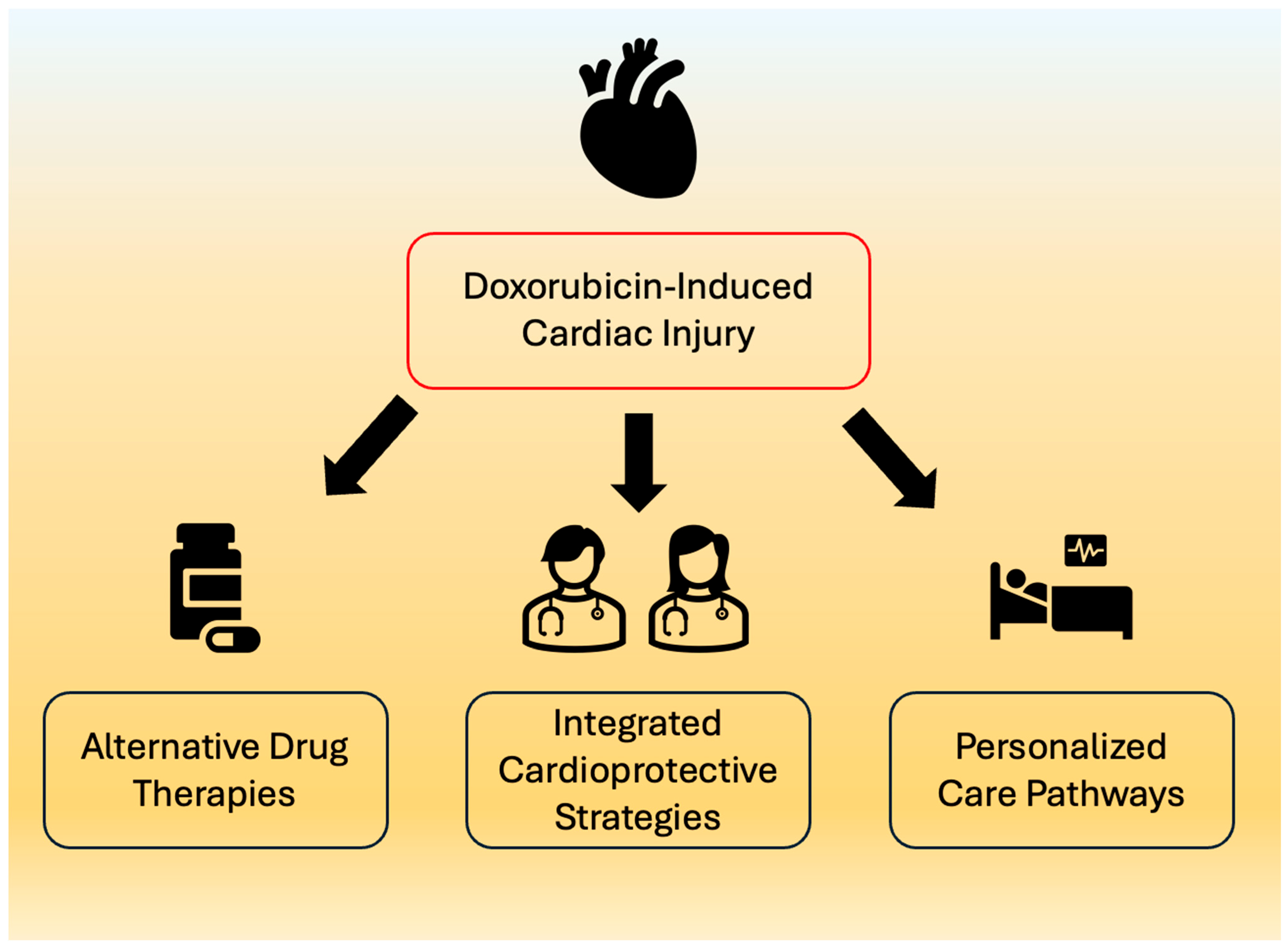
16. Cardioprotective Strategies and Interventions
17. Monitoring Guidelines and Risk Stratification
18. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghigo, A.; Ameri, P.; Asnani, A.; Bertero, E.; de Boer, R.A.; Farmakis, D.; Gonzalez, A.; Heymans, S.; Ibanez, B.; Lopez-Fernandez, T.; et al. Update on preclinical models of cancer therapy-related cardiac dysfunction: Challenges and perspectives. A scientific statement of the Heart Failure Association (HFA) of the ESC, the ESC Council of Cardio-Oncology, and the ESC Working Group on Cellular Biology of the Heart. Eur. J. Heart Fail. 2025; early view. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, L.; Chen, L.; Wang, X. Doxorubicin-Induced Cardiac Remodeling: Mechanisms and Mitigation Strategies. Cardiovasc. Drugs Ther. 2025. [Google Scholar] [CrossRef]
- Cejas, R.B.; Petrykey, K.; Sapkota, Y.; Burridge, P.W. Anthracycline Toxicity: Light at the End of the Tunnel? Annu. Rev. Pharmacol. Toxicol. 2024, 64, 115–134. [Google Scholar] [CrossRef]
- Linders, A.N.; Dias, I.B.; Lopez Fernandez, T.; Tocchetti, C.G.; Bomer, N.; Van der Meer, P. A review of the pathophysiological mechanisms of doxorubicin-induced cardiotoxicity and aging. npj Aging 2024, 10, 9. [Google Scholar] [CrossRef]
- Salloum, F.N.; Tocchetti, C.G.; Ameri, P.; Ardehali, H.; Asnani, A.; de Boer, R.A.; Burridge, P.; Cabrera, J.A.; de Castro, J.; Cordoba, R.; et al. Priorities in Cardio-Oncology Basic and Translational Science: GCOS 2023 Symposium Proceedings: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2023, 5, 715–731. [Google Scholar] [CrossRef]
- Nicoletto, R.E.; Ofner, C.M., 3rd. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Zielinski, R.; Winkler, C.; Silberman, S.; Reuther, S.; Priebe, W. Anthracycline-induced cardiotoxicity—Are we about to clear this hurdle? Eur. J. Cancer 2023, 185, 94–104. [Google Scholar] [CrossRef]
- Zaheed, M.; Wilcken, N.; Willson, M.L.; O’Connell, D.L.; Goodwin, A. Sequencing of anthracyclines and taxanes in neoadjuvant and adjuvant therapy for early breast cancer. Cochrane Database Syst. Rev. 2019, 2, CD012873. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Anthracycline-containing and taxane-containing chemotherapy for early-stage operable breast cancer: A patient-level meta-analysis of 100 000 women from 86 randomised trials. Lancet 2023, 401, 1277–1292. [Google Scholar] [CrossRef]
- Balough, E.; Ariza, A.; Asnani, A.; Hoeger, C.W. Cardiotoxicity of Anthracyclines. Cardiol. Clin. 2025, 43, 111–127. [Google Scholar] [CrossRef]
- Lohr, D.; Thiele, A.; Stahnke, M.; Braun, V.M.; Klopfleisch, R.; Klein, O.; Dresen, S.; Landmesser, U.; Foryst-Ludwig, A.; Kintscher, U.; et al. Characterization of anthracycline-induced cardiotoxicity by diffusion tensor magnetic resonance imaging. Basic. Res. Cardiol. 2025, 120, 57–69. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef]
- Omland, T.; Heck, S.L.; Gulati, G. The Role of Cardioprotection in Cancer Therapy Cardiotoxicity: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 19–37. [Google Scholar] [CrossRef]
- Narezkina, A.; Nasim, K. Anthracycline Cardiotoxicity. Circ. Heart Fail. 2019, 12, e005910. [Google Scholar] [CrossRef]
- Willis, M.S.; Parry, T.L.; Brown, D.I.; Mota, R.I.; Huang, W.; Beak, J.Y.; Sola, M.; Zhou, C.; Hicks, S.T.; Caughey, M.C.; et al. Doxorubicin Exposure Causes Subacute Cardiac Atrophy Dependent on the Striated Muscle-Specific Ubiquitin Ligase MuRF1. Circ. Heart Fail. 2019, 12, e005234. [Google Scholar] [CrossRef]
- Camilli, M.; Maggio, L.; Tinti, L.; Torre, I.; Viscovo, M.; Viscovo, M.; Tamburrini, G.; Lombardo, A.; Cardinale, D.M.; Minotti, G.; et al. Cardio-oncology: Emerging Concepts in Cardiovascular Sequelae of Cancer Therapies, Translational Research and Reverse Cardio-oncology. Eur. Cardiol. 2025, 20, e05. [Google Scholar] [CrossRef]
- Serres, S.; Tardin, C.; Salomé, L. Single-Molecule Sensing of DNA Intercalating Drugs in Water. Anal. Chem. 2020, 92, 8151–8158. [Google Scholar] [CrossRef]
- Taymaz-Nikerel, H.; Karabekmez, M.E.; Eraslan, S.; Kırdar, B. Doxorubicin Induces an Extensive Transcriptional and Metabolic Rewiring in Yeast Cells. Sci. Rep. 2018, 8, 13672. [Google Scholar] [CrossRef]
- Hari, A.D.; Naidu, V.G.M.; Das, U.N. N-6 and N-3 Fatty Acids and Their Metabolites Augment Inhibitory Action of Doxorubicin on the Proliferation of Human Neuroblastoma (IMR-32) Cells by Enhancing Lipid Peroxidation and Suppressing Ras, Myc, and Fos. Biofactors 2018, 44, 387–401. [Google Scholar] [CrossRef]
- Saroj, N.; Dholaniya, P.S.; Alvi, S.B.; Sridharan, D.; Soni, N.; Ashraf, S.A.; Choudhry, A.; Ashraf, Y.A.; Mikula, S.K.; Singla, D.K.; et al. SiRNA-mediated knockdown of TOP2B protects hiPSC-derived cardiomyocytes from doxorubicin-induced toxicity. Life Sci. 2025, 371, 123595. [Google Scholar] [CrossRef]
- Korga-Plewko, A.; Ostrowska, M.; Józefczyk, A.; Iwan, M.; Wójcik, R.; Zgórka, G.; Herbet, M.; Vilarrubla, G.G.; Dudka, J. Apigenin and Hesperidin Augment the Toxic Effect of Doxorubicin Against HepG2 Cells. BMC Pharmacol. Toxicol. 2019, 20, 22. [Google Scholar] [CrossRef]
- Du, J.; Zhang, A.; Li, J.; Liu, X.; Wu, S.; Wang, B.; Wang, Y.; Jia, H. Doxorubicin-Induced Cognitive Impairment: The Mechanistic Insights. Front. Oncol. 2021, 11, 673340. [Google Scholar] [CrossRef]
- Yan, M.; Cao, Y.; Wang, Q.; Xu, K.; Dou, L.; Huang, X.; Chen, B.; Tang, W.; Lan, M.; Liu, B.; et al. miR-488-3p Protects Cardiomyocytes Against Doxorubicin-Induced Cardiotoxicity by Inhibiting CyclinG1. Oxidative Med. Cell. Longev. 2022, 2022, 5184135. [Google Scholar] [CrossRef] [PubMed]
- Negm, A.; Mersal, E.A.; Dawood, A.F.; Abd El-Azim, A.O.; Hasan, O.; Alaqidi, R.; Alotaibi, A.; Alshahrani, M.; Alheraiz, A.; Shawky, T.M. Multifaceted Cardioprotective Potential of Reduced Glutathione Against Doxorubicin-Induced Cardiotoxicity via Modulating Inflammation-Oxidative Stress Axis. Int. J. Mol. Sci. 2025, 26, 3201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Wang, R.; Yi, Q.; Xu, H.; Tan, B.; Zhu, J. Kartogenin Improves Doxorubicin-Induced Cardiotoxicity by Alleviating Oxidative Stress and Protecting Mitochondria. Int. J. Mol. Sci. 2025, 26, 2434. [Google Scholar] [CrossRef]
- Lai, C.; Cole, D.E.; Steinberg, S.M.; Lucas, N.; Dombi, E.; Melani, C.; Roschewski, M.; Balis, F.M.; Widemann, B.C.; Wilson, W.H. Doxorubicin Pharmacokinetics and Toxicity in Patients With Aggressive Lymphoma and Hepatic Impairment. Blood Adv. 2023, 7, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Wen, Z.; Meng, J.; Yang, Y.; Zhang, Y.; Wang, J.; Cao, X. Comparative Pharmacokinetics of Free Doxorubicin and a Liposomal Formulation in Cats Following Intravenous Administration. Front. Vet. Sci. 2024, 11, 1353775. [Google Scholar] [CrossRef]
- Gándara-Mireles, J.A.; Lares-Asseff, I.; Espinoza, E.A.R.; Fierro, I.V.; Castañeda, V.L.; Hurtado, L.P.C.; González, C.D.; Romero, L.P.; Almanza-Reyes, H. Impact of Single-Nucleotide Variants and Nutritional Status on Population Pharmacokinetics of Doxorubicin, and Its Effect on Cardiotoxicity in Children With Leukemia. J. Oncol. Pharm. Pract. 2022, 29, 1290–1305. [Google Scholar] [CrossRef]
- Dragojevic, S.; Turner, L.; Raucher, D. Circumventing Doxorubicin Resistance Using Elastin-Like Polypeptide Biopolymer-Mediated Drug Delivery. Int. J. Mol. Sci. 2022, 23, 2301. [Google Scholar] [CrossRef]
- Islam, M.R.; Patel, J.; Back, P.I.; Shmeeda, H.; Adamsky, K.; Yang, H.; Álvarez, C.; Gabizón, A.; La-Beck, N.M. Comparative Effects of Free Doxorubicin, Liposome Encapsulated Doxorubicin and Liposome Co-Encapsulated Alendronate and Doxorubicin (PLAD) on the Tumor Immunologic Milieu in a Mouse Fibrosarcoma Model. Nanotheranostics 2022, 6, 451–464. [Google Scholar] [CrossRef]
- Liu, Y.; Corrales-Guerrero, S.; Kuo, J.C.; Robb, R.; Nagy, G.; Cui, T.; Lee, R.J.; Williams, T.M. Improved Targeting and Safety of Doxorubicin Through a Novel Albumin Binding Prodrug Approach. ACS Omega 2023, 9, 977–987. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Wang, P.; Zhou, Q.; Zhang, K.; Zhang, J.; Tian, J. Nonclinical Study of the Active Components of Doxorubicin Hydrochloride Liposome Injection in Vivo. Pharmacol. Pharm. 2023, 14, 363–375. [Google Scholar] [CrossRef]
- Maulik, A.; Davidson, S.M.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Ischaemic Preconditioning Protects Cardiomyocytes From Anthracycline-Induced Toxicity via the PI3K Pathway. Cardiovasc. Drugs Ther. 2018, 32, 245–253. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Xie, B.; Liu, D.; Wang, Y.; Zhou, Z.; Zhang, Y.; King, E.V.; Tse, G.; Liu, T. Resveratrol Activation of SIRT1/MFN2 Can Improve Mitochondria Function, Alleviating Doxorubicin-induced Myocardial Injury. Cancer Innov. 2023, 2, 253–264. [Google Scholar] [CrossRef]
- Dong, L.; Liu, H. Triptonide Protects Against Doxorubicin-Induced Cardiotoxicity in Rats by Regulating Oxidative Stress and Cardiac Biomarkers. Indian J. Pharm. Educ. Res. 2023, 57, 787–796. [Google Scholar] [CrossRef]
- Benzer, F.; Kandemir, F.M.; Özkaraca, M.; Küçükler, S.; Çağlayan, C. Curcumin Ameliorates Doxorubicin-induced Cardiotoxicity by Abrogation of Inflammation, Apoptosis, Oxidative DNA Damage, and Protein Oxidation in Rats. J. Biochem. Mol. Toxicol. 2018, 32, e22030. [Google Scholar] [CrossRef]
- Tanaka, R.; Umemura, M.; Narikawa, M.; Hikichi, M.; Osaw, K.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Tamura, K.; Ishikawa, Y. Reactive Fibrosis Precedes Doxorubicin-induced Heart Failure Through Sterile Inflammation. Esc. Heart Fail. 2020, 7, 588–603. [Google Scholar] [CrossRef]
- Li, Z.; Ye, Z.; Ma, J.; Gu, Q.; Teng, J.; Gong, X. MicroRNA-133b Alleviates Doxorubicin-induced Cardiomyocyte Apoptosis and Cardiac Fibrosis by Targeting PTBP1 and TAGLN2. Int. J. Mol. Med. 2021, 48, 125. [Google Scholar] [CrossRef]
- Subbarao, R.B.; Ok, S.H.; Lee, S.H.; Kang, D.; Kim, E.J.; Kim, J.Y.; Sohn, J.T. Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts. Cells 2018, 7, 144. [Google Scholar] [CrossRef]
- Yu, W.; Qin, X.; Zhang, Y.; Qiu, P.; Wang, L.; Zha, W.; Ren, J. Curcumin Suppresses Doxorubicin-Induced Cardiomyocyte Pyroptosis via a PI3K/Akt/mTOR-dependent Manner. Cardiovasc. Diagn. Ther. 2020, 10, 752–769. [Google Scholar] [CrossRef]
- Bai, X.; Wei, H.; Liu, G.; Li, L. Astragalus polyphenols attenuates doxorubicin-induced cardiotoxicity by activating the PI3K/AKT/NRF2 pathway. PLoS ONE 2025, 20, e0319067. [Google Scholar] [CrossRef]
- Hayek, E.R.; Speakman, E.; Rehmus, E. Acute doxorubicin cardiotoxicity. N. Engl. J. Med. 2005, 352, 2456–2457. [Google Scholar] [CrossRef]
- Kariuki, N.; Kimani, E.; Jowi, C.; Wamalwa, D.; Suen, J.Y.; Fraser, J.F.; Obonyo, N.G. Early myocardial injury in children on doxorubicin for cancer chemotherapy: A cross-sectional study in a tertiary referral centre in Kenya. BMC Cardiovasc. Disord. 2024, 24, 260. [Google Scholar] [CrossRef]
- Dulf, P.L.; Mocan, M.; Coada, C.A.; Dulf, D.V.; Moldovan, R.; Baldea, I.; Farcas, A.D.; Blendea, D.; Filip, A.G. Doxorubicin-induced acute cardiotoxicity is associated with increased oxidative stress, autophagy, and inflammation in a murine model. Naunyn Schmiedebergs Arch. Pharmacol. 2023, 396, 1105–1115. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Belger, C.; Abrahams, C.; Imamdin, A.; Lecour, S. Doxorubicin-induced cardiotoxicity and risk factors. Int. J. Cardiol. Heart Vasc. 2024, 50, 101332. [Google Scholar] [CrossRef]
- Wang, F.; Chandra, J.; Kleinerman, E.S. Exercise intervention decreases acute and late doxorubicin-induced cardiotoxicity. Cancer Med. 2021, 10, 7572–7584. [Google Scholar] [CrossRef]
- Varghese, S.S.; Eekhoudt, C.R.; Jassal, D.S. Mechanisms of anthracycline-mediated cardiotoxicity and preventative strategies in women with breast cancer. Mol. Cell Biochem. 2021, 476, 3099–3109. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Ma, W.; Wei, S.; Zhang, B.; Li, W. Molecular Mechanisms of Cardiomyocyte Death in Drug-Induced Cardiotoxicity. Front. Cell Dev. Biol. 2020, 8, 434. [Google Scholar] [CrossRef]
- Pharoah, B.M.; Zhang, C.; Khodade, V.S.; Keceli, G.; McGinity, C.; Paolocci, N.; Toscano, J.P. Hydropersulfides (RSSH) attenuate doxorubicin-induced cardiotoxicity while boosting its anticancer action. Redox Biol. 2023, 60, 102625. [Google Scholar] [CrossRef]
- Belmonte, F.; Das, S.; Sysa-Shah, P.; Sivakumaran, V.; Stanley, B.; Guo, X.; Paolocci, N.; Aon, M.A.; Nagane, M.; Kuppusamy, P.; et al. ErbB2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1271–H1280. [Google Scholar] [CrossRef]
- Ikegami, E.; Fukazawa, R.; Kanbe, M.; Watanabe, M.; Abe, M.; Watanabe, M.; Kamisago, M.; Hajikano, M.; Katsube, Y.; Ogawa, S. Edaravone, a potent free radical scavenger, prevents anthracycline-induced myocardial cell death. Circ. J. 2007, 71, 1815–1820. [Google Scholar] [CrossRef]
- Li, K.; Sung, R.Y.; Huang, W.Z.; Yang, M.; Pong, N.H.; Lee, S.M.; Chan, W.Y.; Zhao, H.; To, M.Y.; Fok, T.F.; et al. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation 2006, 113, 2211–2220. [Google Scholar] [CrossRef]
- Dhingra, R.; Margulets, V.; Chowdhury, S.R.; Thliveris, J.; Jassal, D.; Fernyhough, P.; Dorn, G.W., 2nd; Kirshenbaum, L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E5537–E5544. [Google Scholar] [CrossRef]
- Sysa-Shah, P.; Tocchetti, C.G.; Gupta, M.; Rainer, P.P.; Shen, X.; Kang, B.H.; Belmonte, F.; Li, J.; Xu, Y.; Guo, X.; et al. Bidirectional cross-regulation between ErbB2 and beta-adrenergic signalling pathways. Cardiovasc. Res. 2016, 109, 358–373. [Google Scholar] [CrossRef]
- Amgalan, D.; Garner, T.P.; Pekson, R.; Jia, X.F.; Yanamandala, M.; Paulino, V.; Liang, F.G.; Corbalan, J.J.; Lee, J.; Chen, Y.; et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat. Cancer 2020, 1, 315–328. [Google Scholar] [CrossRef]
- Tungalag, T.; Kang, H.S.; Yang, D.K. Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway. Antioxidants 2025, 14, 337. [Google Scholar] [CrossRef]
- Xu, Y.; Sui, Y.; Jiang, R.; Wang, X.; Suda, M.; Niimi, M.; Mao, Z.; Zhang, Z.; Zhang, S.L.; Fan, J.; et al. Sulfhydrated albumin transmits H(2)S signaling and ameliorates DOX-induced multiorgan injuries. Redox Biol. 2025, 83, 103631. [Google Scholar] [CrossRef]
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.L.; Gupta, M.P. Honokiol, an Activator of Sirtuin-3 (SIRT3) Preserves Mitochondria and Protects the Heart From Doxorubicin-Induced Cardiomyopathy in Mice. Oncotarget 2017, 8, 34082–34098. [Google Scholar] [CrossRef]
- Lim, C.C.; Zuppinger, C.; Guo, X.; Kuster, G.M.; Helmes, M.; Eppenberger, H.M.; Suter, T.M.; Liao, R.; Sawyer, D.B. Anthracyclines Induce Calpain-Dependent Titin Proteolysis and Necrosis in Cardiomyocytes. J. Biol. Chem. 2004, 279, 8290–8299. [Google Scholar] [CrossRef]
- Hafez, A.A.; Jamali, Z.; Samiei, S.; Khezri, S.; Salimi, A. Reduction of Doxorubicin-Induced Cytotoxicity and Mitochondrial Damage by Betanin in Rat Isolated Cardiomyocytes and Mitochondria. Human. Exp. Toxicol. 2021, 40, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wan, Q.; Li, Q.; Fang, J.; Peng, L.; Hu, J. Substance P Prevents Doxorubicin-induced Cardiomyocyte Injury by Regulating Apoptosis and Autophagy: In vitro and in vivo Evidence. Mol. Med. Rep. 2021, 25, 50. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, H.; Chen, H.; Ren, Y.; Li, J.; Gong, L.; Zhong, L.; Yang, J. Doxorubicin-induced apoptosis is exacerbated by MG53 and associated with altered Akt signaling in H9c2 cells. Mol. Pharmacol. 2025, 107, 100032. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, T.; Huang, X.; Lin, Y.; Chen, B.; Pang, J.; Li, G.; Wang, Q.; Zohrabian, S.; Duan, C.; et al. Astragalus Polysaccharide Restores Autophagic Flux and Improves Cardiomyocyte Function in Doxorubicin-Induced Cardiotoxicity. Oncotarget 2016, 8, 4837–4848. [Google Scholar] [CrossRef]
- Huang, K.C.; Kuo, W.-W.; Shen, C.-Y.; Chen, Y.; Lin, Y.-M.; Ho, T.J.; Padma, V.V.; Lo, J.F.; Huang, C.Y. Anthocyanin Attenuates Doxorubicin-Induced Cardiomyotoxicity via Estrogen Receptor-A/Β and Stabilizes HSF1 to Inhibit the IGF-IIR Apoptotic Pathway. Int. J. Mol. Sci. 2016, 17, 1588. [Google Scholar] [CrossRef] [PubMed]
- Kumari, H.; Huang, W.-H.; Chan, M.W. Review on the Role of Epigenetic Modifications in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 2020, 7, 56. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin Induces Cardiotoxicity Through Upregulation of Death Receptors Mediated Apoptosis in Cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef]
- Zou, R.; Wang, S.; Hong, C.; Wang, Y.; Wang, C. Pharmacological Activation of Rev-Erbα Attenuates Doxorubicin-Induced Cardiotoxicity by PGC-1α Signaling Pathway. Cardiovasc. Ther. 2023, 2023, 2108584. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, W.; Zhang, M.; Jin, M.; Xu, W.; Zhou, X. Gas Signaling Molecule Hydrogen Sulfide Attenuates Doxorubicin-Induced Dilated Cardiomyopathy. Oncotarget 2017, 8, 95425–95431. [Google Scholar] [CrossRef]
- Han, X.; Ren, D.M.; Fan, P.; Shen, T.; Lou, H.X. Protective Effects of Naringenin-7-O-Glucoside on Doxorubicin-Induced Apoptosis in H9C2 Cells. Eur. J. Pharmacol. 2008, 581, 47–53. [Google Scholar] [CrossRef]
- Shi, X.; Xu, J.; Zhong, X.; Qian, Y.; Lin, L.; Fang, Z.; Ye, B.; Lyu, Y.; Zhang, R.; Zheng, Z.; et al. Deubiquitinase MYSM1 promotes doxorubicin-induced cardiotoxicity by mediating TRIM21-ferroptosis axis in cardiomyocytes. Cell Commun. Signal. 2024, 22, 593. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhu, M.; Xu, X.; Li, X.; Yao, Y.; Liu, C.; He, K. AMPD3 promotes doxorubicin-induced cardiomyopathy through HSP90alpha-mediated ferroptosis. iScience 2024, 27, 111005, Erratum in iScience 2024, 27, 111304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Wang, J.; Li, Z.; Wang, S.; Yang, W.; Hai, Y.; Liu, D. Kaempferol Protects against Doxorubicin-Induced Myocardial Damage by Inhibiting Mitochondrial ROS-Dependent Ferroptosis. Redox Rep. 2025, 30, 2503130. [Google Scholar] [CrossRef]
- Hou, Y.; Gao, W.; Lui, K.O. A hidden role of Th17 cells in doxorubicin-induced cardiac ferroptosis. Cardiovasc. Res. 2024, 120, 1989–1991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, W.; Yin, Z.; Liu, S.; Zhao, M.; Xu, Y.; Liu, J.; Pan, W.; Peng, S.; Wei, C.; et al. Interleukin-12p40 deficiency attenuates myocardial ferroptosis in doxorubicin-induced chronic cardiomyopathy by inhibiting Th17 differentiation and interleukin-17A production. Cardiovasc. Res. 2024, 120, 2117–2133. [Google Scholar] [CrossRef]
- Wu, L.; Wang, L.T.; Du, Y.X.; Zhang, Y.M.; Ren, J. Asiatic acid ameliorates doxorubicin-induced cardiotoxicity by promoting FPN-mediated iron export and inhibiting ferroptosis. Acta Pharmacol. Sin. 2025, 46, 81–95. [Google Scholar] [CrossRef]
- Liu, D.; Cheng, X.; Wu, H.; Song, H.; Bu, Y.; Wang, J.; Zhang, X.; Yan, C.; Han, Y. CREG1 attenuates doxorubicin-induced cardiotoxicity by inhibiting the ferroptosis of cardiomyocytes. Redox Biol. 2024, 75, 103293. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Wang, G.; Ren, J. Molecular Mechanisms and Therapeutic Targeting of Ferroptosis in Doxorubicin-Induced Cardiotoxicity. JACC Basic. Transl. Sci. 2024, 9, 811–826. [Google Scholar] [CrossRef]
- El-Gohary, R.M.; Okasha, A.H.; Abd El-Azeem, A.H.; Abdel Ghafar, M.T.; Ibrahim, S.; Hegab, I.I.; Farghal, E.E.; Shalaby, S.A.F.; Elshora, O.A.; ElMehy, A.E.; et al. Uncovering the Cardioprotective Potential of Diacerein in Doxorubicin Cardiotoxicity: Mitigating Ferritinophagy-Mediated Ferroptosis via Upregulating NRF2/SLC7A11/GPX4 Axis. Antioxidants 2024, 13, 493. [Google Scholar] [CrossRef]
- Wu, L.; Du, Y.; Wang, L.; Zhang, Y.; Ren, J. Inhibition of METTL3 ameliorates doxorubicin-induced cardiotoxicity through suppression of TFRC-mediated ferroptosis. Redox Biol. 2024, 72, 103157. [Google Scholar] [CrossRef]
- Zhai, Y.; Bai, J.; Peng, Y.; Cao, J.; Fang, G.; Dong, Y.; Wang, Z.; Lu, Y.; Wang, M.; Liu, M.; et al. Ginsenoside Rb1 attenuates doxorubicin induced cardiotoxicity by suppressing autophagy and ferroptosis. Biochem. Biophys. Res. Commun. 2024, 710, 149910. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Li, Y.; Xu, X.; Yang, H.; Li, X.; Fu, S.; Guo, Z.; Zhang, J.; Li, H.; Tian, J. Neutrophil extracellular traps mediate cardiomyocyte ferroptosis via the Hippo-Yap pathway to exacerbate doxorubicin-induced cardiotoxicity. Cell. Mol. Life Sci. 2024, 81, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qiao, Y.; Yu, J.; Wang, Q.; Wu, X.; Cao, Q.; Zhang, Z.; Feng, Z.; He, H. Endurance exercise preconditioning alleviates ferroptosis induced by doxorubicin-induced cardiotoxicity through mitochondrial superoxide-dependent AMPKalpha2 activation. Redox Biol. 2024, 70, 103079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pan, J.; Huang, S.; Chen, X.; Chang, A.C.Y.; Wang, C.; Zhang, J.; Zhang, H. Hydrogen sulfide protects cardiomyocytes from doxorubicin-induced ferroptosis through the SLC7A11/GSH/GPx4 pathway by Keap1 S-sulfhydration and Nrf2 activation. Redox Biol. 2024, 70, 103066. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, J.; Zhang, J.; Shi, H.; Wang, J.; Yan, Y. FTO ameliorates doxorubicin-induced cardiotoxicity by inhibiting ferroptosis via P53-P21/Nrf2 activation in a HuR-dependent m6A manner. Redox Biol. 2024, 70, 103067. [Google Scholar] [CrossRef]
- Shen, M.; Cao, S.; Long, X.; Xiao, L.; Yang, L.; Zhang, P.; Li, L.; Chen, F.; Lei, T.; Gao, H.; et al. DNAJC12 causes breast cancer chemotherapy resistance by repressing doxorubicin-induced ferroptosis and apoptosis via activation of AKT. Redox Biol. 2024, 70, 103035. [Google Scholar] [CrossRef]
- Chen, G.; Luo, S.; Guo, H.; Lin, J.; Xu, S. Licochalcone A alleviates ferroptosis in doxorubicin-induced cardiotoxicity via the PI3K/AKT/MDM2/p53 pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 4247–4262. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, W.; Yin, L.; Huang, C.; Zhao, H. Exogenous Maresin1 attenuates doxorubicin-induced cardiomyocyte ferroptosis and mitochondrial impairment via NRF2/GPX4 axis. Free Radic. Biol. Med. 2025, 235, 335–346. [Google Scholar] [CrossRef]
- Cui, L.; Xia, Q.; Wang, Y.; Han, C.; Zang, X.; Zhang, L.; Xing, J.; Zheng, R.; Zhang, Y. Luteolin-7-O-glucuronide alleviates doxorubicin-induced cardiotoxicity by inhibiting PPAR-mediated ferroptosis. Toxicol. Appl. Pharmacol. 2025, 500, 117381. [Google Scholar] [CrossRef]
- Yu, J.; Wang, J.; Liu, X.; Wang, C.; Wu, L.; Zhang, Y. Bioinformatics analysis of ferroptosis-related biomarkers and potential drug predictions in doxorubicin-induced cardiotoxicity. Front. Cardiovasc. Med. 2025, 12, 1566782. [Google Scholar] [CrossRef]
- Li, J.; Zeng, Y.; Liu, F.; Liao, X.; Zhong, C.; Dong, S.; Cai, Y.; Yang, P. Erythrocyte Membrane-Camouflaged Xanthohumol Nanoparticles Mitigate Doxorubicin-Induced Cardiotoxicity by Inhibiting Ferroptosis. ACS Biomater. Sci. Eng. 2025, 11, 2727–2738. [Google Scholar] [CrossRef]
- Shi, Y.; Cai, J.; Chen, L.; Cheng, H.; Song, X.; Xue, J.; Xu, R.; Ma, J.; Ge, J. AIG1 protects against doxorubicin-induced cardiomyocyte ferroptosis and cardiotoxicity by promoting ubiquitination-mediated p53 degradation. Theranostics 2025, 15, 4931–4954. [Google Scholar] [CrossRef]
- Cao, X.; Zhao, L.; Zhou, J.; Ding, S.; Sun, Y.; Ma, Y.; Ma, Z.; Liu, H.; Dong, T.; Luo, A.; et al. Dexmedetomidine inhibits ferroptosis through the Akt/GSK3beta/Nrf2 axis and alleviates adriamycin-induced cardiotoxicity. Life Sci. 2025, 371, 123609. [Google Scholar] [CrossRef]
- Al-kuraishy, H.M. Cardio-Protective Effects of Cyclosporine in Doxorubicin Induced Cardiotoxicity and Assessment of Interleukin-17 as Biomarker of Cardiac Injury an Animal Model Study. Adv. Biomed. Pharm. 2015, 2, 138–145. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, X.; Tan, B.; Qing-ya, Z.; Zhao, X.; Dong, D. Potential Role of Endoplasmic Reticulum Stress in Doxorubicin-Induced Cardiotoxicity-an Update. Front. Pharmacol. 2024, 15, 1415108. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, L. In Vitro and in Vivo Cardioprotective Effects of Curcumin Against Doxorubicin-Induced Cardiotoxicity: A Systematic Review. J. Oncol. 2022, 2022, 7277562. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Guo, X.; Tao, Z.; Wu, C.; Jiang, M.; Pu, J. Empagliflozin attenuates DOX-induced cardiotoxicity by inhibiting RIPK1-mediated endoplasmic reticulum stress and autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167898. [Google Scholar] [CrossRef]
- Al-Maamari, A.; Sultan, M.; Wu, S.; Zhang, T.; Wang, C.; Han, B.; Duan, Y.; Ding, S.S.; Chen, N.; Zhang, H.; et al. Activation of sigma 1 receptor attenuates doxorubicin-induced cardiotoxicity by alleviating oxidative stress, mitochondria dysfunction, ER stress-related apoptosis, and autophagy impairment. Int. J. Biol. Macromol. 2025, 310, 143549. [Google Scholar] [CrossRef]
- Elariny, H.A.; Atia, H.A.; Abdallah, M.H.; Khalifa, A.M.; Abd Elmaaboud, M.A.; Elkady, M.A.; Kabel, A.M. Olmesartan attenuates doxorubicin-elicited testicular toxicity: The interaction between sirtuin-1, HMGB1/NLRP3 inflammasome/gasdermin D signaling, and AMPK/mTOR-driven autophagy. Life Sci. 2025, 370, 123545. [Google Scholar] [CrossRef]
- Tan, N.; Luo, H.; Li, W.; Ling, G.; Wei, Y.; Wang, W.; Wang, Y. The dual function of autophagy in doxorubicin-induced cardiotoxicity: Mechanism and natural products. Semin. Cancer Biol. 2025, 109, 83–90. [Google Scholar] [CrossRef]
- Hu, F.; Yan, S.; Lin, L.; Qiu, X.; Lin, X.; Wang, W. Sacubitril/valsartan attenuated myocardial inflammation, fibrosis, apoptosis and promoted autophagy in doxorubicin-induced cardiotoxicity mice via regulating the AMPKalpha-mTORC1 signaling pathway. Mol. Cell. Biochem. 2025, 480, 1891–1908. [Google Scholar] [CrossRef]
- Liao, C.C.; Long, Y.; Tsai, M.L.; Lin, C.Y.; Hsu, K.W.; Lee, C.H. G-cleave LC3B biosensor: Monitoring autophagy and assessing resveratrol’s synergistic impact on doxorubicin-induced apoptosis in breast cancer cells. Breast Cancer Res. 2024, 26, 190. [Google Scholar] [CrossRef]
- Guan, J.; Mo, H.; Virak, V.; Guo, R.; Que, D.; Yu, W.; Zhang, X.; Yan, J.; Wang, Y.; Yang, Y.; et al. eEF2K alleviates doxorubicin-induced cardiotoxicity by inhibiting GSK3beta and improving autophagy dysfunction. Cell Biol. Toxicol. 2024, 41, 15. [Google Scholar] [CrossRef]
- Zhu, H.; Jiang, C.W.; Zhang, W.L.; Yang, Z.Y.; Sun, G. Targeting oncogenic MAGEA6 sensitizes triple negative breast cancer to doxorubicin through its autophagy and ferroptosis by stabling AMPKalpha1. Cell Death Discov. 2024, 10, 430. [Google Scholar] [CrossRef]
- Ou, W.; Liu, H.; Chen, C.; Yang, C.; Zhao, X.; Zhang, Y.; Zhang, Z.; Huang, S.; Mo, H.; Lu, W.; et al. Spexin inhibits excessive autophagy-induced ferroptosis to alleviate doxorubicin-induced cardiotoxicity by upregulating Beclin 1. Br. J. Pharmacol. 2024, 181, 4195–4213. [Google Scholar] [CrossRef]
- Bientinesi, E.; Ristori, S.; Lulli, M.; Monti, D. Quercetin induces senolysis of doxorubicin-induced senescent fibroblasts by reducing autophagy, preventing their pro-tumour effect on osteosarcoma cells. Mech. Ageing Dev. 2024, 220, 111957. [Google Scholar] [CrossRef]
- Lin, Z.H.; Xiang, H.Q.; Yu, Y.W.; Xue, Y.J.; Wu, C.; Lin, C.; Ji, K.T. Dihydroartemisinin alleviates doxorubicin-induced cardiotoxicity and ferroptosis by activating Nrf2 and regulating autophagy. FASEB J. 2024, 38, e23677. [Google Scholar] [CrossRef]
- Ma, Y.; Zhao, H.-P.; Yang, L.-G.; Li, L.; Wang, A.-L.; Zhang, X.-J.; Wang, K.; Yang, B.; Zhu, Z.-F.; Zhang, P.-J.; et al. NADPH oxidase 2 mediates cardiac sympathetic denervation and myocyte autophagy, resulting in cardiac atrophy and dysfunction in doxorubicin-induced cardiomyopathy. Sci. Rep. 2024, 14, 6971. [Google Scholar] [CrossRef]
- Shuey, A.; Patricelli, C.; Oxford, J.T.; Pu, X. Effects of doxorubicin on autophagy in fibroblasts. Hum. Exp. Toxicol. 2024, 43, 9603271241231947. [Google Scholar] [CrossRef]
- Zhao, H.P.; Ma, Y.; Zhang, X.J.; Guo, H.X.; Yang, B.; Chi, R.F.; Zhang, N.P.; Wang, J.P.; Li, B.; Qin, F.Z.; et al. NADPH oxidase 2 inhibitor GSK2795039 prevents doxorubicin-induced cardiac atrophy by attenuating cardiac sympathetic nerve terminal abnormalities and myocyte autophagy. Eur. J. Pharmacol. 2024, 967, 176351. [Google Scholar] [CrossRef]
- Jin, W.; Yang, T.; Jia, J.; Jia, J.; Zhou, X. Enhanced Sensitivity of A549 Cells to Doxorubicin with WS(2) and WSe(2) Nanosheets via the Induction of Autophagy. Int. J. Mol. Sci. 2024, 25, 1164. [Google Scholar] [CrossRef]
- Hosseini, A.; Safari, M.-K.; Rajabian, A.; Boroumand-Noughabi, S.; Eid, A.H.; Dhaheri, Y.A.; Gumpricht, E.; Sahebkar, A. Cardioprotective Effect of Rheum Turkestanicum Against Doxorubicin-Induced Toxicity in Rats. Front. Pharmacol. 2022, 13, 909079. [Google Scholar] [CrossRef]
- Khafaji, A.T.A.; Barakat, A.; Shayyal, A.J.; Taan, A.A.; Al-Aouadi, R.F.A. Managing Doxorubicin Cardiotoxicity: Insights Into Molecular Mechanisms and Protective Strategies. J. Biochem. Mol. Toxicol. 2025, 39, e70155. [Google Scholar] [CrossRef]
- Huyan, Y.; Chen, X.; Chang, Y.; Hua, X.; Fan, X.; Shan, D.; Xu, Z.; Tao, M.; Zhang, H.; Liu, S.; et al. Single-Cell Transcriptomic Analysis Reveals Myocardial Fibrosis Mechanism of Doxorubicin-Induced Cardiotoxicity. Int. Heart J. 2024, 65, 487–497. [Google Scholar] [CrossRef]
- He, W.; Wang, J.; He, W.; Zeng, L.; Zhao, R.; Qiu, K.; Tong, G.; Sun, Z.; He, P. PGAM5 aggravated doxorubicin-induced cardiotoxicity by disturbing mitochondrial dynamics and exacerbating cardiomyocytes apoptosis. Free Radic. Biol. Med. 2025, 235, 95–108. [Google Scholar] [CrossRef]
- Fan, R.; Wang, Y.; Zhang, J.; An, X.; Liu, S.; Bai, J.; Li, J.; Lin, Q.Y.; Xie, Y.; Liao, J.; et al. Hyperhomocysteinaemia Promotes Doxorubicin-Induced Cardiotoxicity in Mice. Pharmaceuticals 2023, 16, 1212. [Google Scholar] [CrossRef]
- Lam, P.Y.; Kutchukian, P.; Anand, R.; Imbriglio, J.; Andrews, C.; Padilla, H.; Vohra, A.; Lane, S.; Parker, D.L., Jr.; Cornella Taracido, I.; et al. Cyp1 Inhibition Prevents Doxorubicin-Induced Cardiomyopathy in a Zebrafish Heart-Failure Model. Chembiochem 2020, 21, 1905–1910. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, C.; Liu, C.; Feng, W. Luteolin Attenuates Doxorubicin-Induced Cardiotoxicity by Modulating the PHLPP1/AKT/Bcl-2 Signalling Pathway. PeerJ 2020, 8, e8845. [Google Scholar] [CrossRef]
- Yang, X.; Liu, S.; Liu, M.; Lou, D.; Zou, W.; Li, X. Trillin protects against doxorubicin-induced cardiotoxicity through regulating Nrf2/HO-1 signaling pathway. PLoS ONE 2025, 20, e0321546. [Google Scholar] [CrossRef]
- Xu, H.-J.; Guo, H.; Tang, Z.; Hao, R.; Wang, S.; Jin, P. Follistatin-like 1 Protects Against Doxorubicin-induced Cardiotoxicity by Preventing Mitochondrial Dysfunction Through the SIRT6/Nrf2 Signaling Pathway. Cell Biol. Int. 2024, 48, 795–807. [Google Scholar] [CrossRef]
- Dutta, B.; Loo, S.; Kam, A.; Wang, X.; Wei, N.; Luo, K.Q.; Liu, C.F.; Tam, J.P. Cell-Permeable Microprotein from Panax Ginseng Protects Against Doxorubicin-Induced Oxidative Stress and Cardiotoxicity. Antioxidants 2025, 14, 493. [Google Scholar] [CrossRef]
- Alyasiry, E.; Janabi, A.; Hadi, N.R. Dipyridamole Ameliorates Doxorubicin-Induced Cardiotoxicity. J. Med. Life 2022, 15, 1184–1190. [Google Scholar] [CrossRef]
- Thonusin, C.; Osataphan, N.; Leemasawat, K.; Nawara, W.; Sriwichaiin, S.; Supakham, S.; Gunaparn, S.; Apaijai, N.; Somwangprasert, A.; Phrommintikul, A.; et al. Changes in Blood Metabolomes as Potential Markers for Severity and Prognosis in Doxorubicin-Induced Cardiotoxicity: A Study in HER2-positive and HER2-negative Breast Cancer Patients. J. Transl. Med. 2024, 22, 398. [Google Scholar] [CrossRef]
- Ding, J.; Feng, X.; Xu, Z.; Xu, H. Metabolomic profiling and biomarker identification for early detection and therapeutic targeting of doxorubicin-induced cardiotoxicity. Front. Cell Dev. Biol. 2025, 13, 1543636. [Google Scholar] [CrossRef]
- Hou, X.; Xie, S.; Zhou, N.; Wei, S.; Yang, Y.; Luo, Z.; Liu, S.; Liu, J.; Xie, N.; Li, W.; et al. Oridonin Alleviates Doxorubicin-Induced Cardiotoxicity by Inhibiting p38 MAPK/MMP3 Signaling Pathway. Chem. Biol. Drug Des. 2025, 105, e70093. [Google Scholar] [CrossRef]
- Yan, Y.; Fang, M.; Zhao, C.; Lin, X.; Tong, C.; Xiang, C.; Ran, Y.; Wang, X.; Li, S.; Chen, G.; et al. Dl-3-n-butylphthalide attenuates DOX-induced cardiotoxicity in mice by inhibiting Nrf2/Keap1 complex formation. Front. Pharmacol. 2025, 16, 1542296. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, S. MiR-27b-3p ameliorates DOX-induced cardiotoxicity by suppressing myocardial inflammation and oxidative stress in mice and cardiomyocytes. Drug Chem. Toxicol. 2025, 1–16. [Google Scholar] [CrossRef]
- El-Refaiy, A.I.; Salem, Z.A.; Badawy, A.A.; Dahran, N.; Desouky, M.A.; El-Magd, M.A. Protective effects of lemon and orange peels and olive oil on doxorubicin-induced myocardial damage via inhibition of oxidative stress and inflammation pathways. Front. Pharmacol. 2025, 16, 1506673. [Google Scholar] [CrossRef]
- Hassen, M.D.; Mousa, N.O.; Radwan, S.M.; Gabre, R.M. The Ameliorative Effect of Interleukin-17A Neutralization on Doxorubicin-Induced Cardiotoxicity by Modulating the NF-kappaB/NLRP3/Caspase-1/IL-1beta Signaling Pathway in Rats. Inflammation 2025. [Google Scholar] [CrossRef]
- Singh, S.K.; Yadav, P.; Patel, D.; Tanwar, S.S.; Sherawat, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Betaine ameliorates doxorubicin-induced cardiomyopathy by inhibiting oxidative stress, inflammation, and fibrosis through the modulation of AMPK/Nrf2/TGF-beta expression. Environ. Toxicol. 2024, 39, 4134–4147. [Google Scholar] [CrossRef]
- Hu, S.; Liu, B.; Yang, M.; Mao, S.; Ju, H.; Liu, Z.; Huang, M.; Wu, G. Carnosic acid protects against doxorubicin-induced cardiotoxicity through enhancing the Nrf2/HO-1 pathway. Food Funct. 2023, 14, 3849–3862. [Google Scholar] [CrossRef]
- Basal, O.A.; Zahran, R.; Saad, E.A. Rifampicin Efficacy Against Doxorubicin-Induced Cardiotoxicity in Mice. Egypt. Heart J. 2023, 75, 73. [Google Scholar] [CrossRef]
- Dorostkar, H.; Haghiralsadat, B.F.; Hemati, M.; Safari, F.; Hassanpour, A.; Naghib, S.M.; Roozbahani, M.H.; Mozafari, M.R.; Moradi, A. Reduction of Doxorubicin-Induced Cardiotoxicity by Co-Administration of Smart Liposomal Doxorubicin and Free Quercetin: In Vitro and in Vivo Studies. Pharmaceutics 2023, 15, 1920. [Google Scholar] [CrossRef]
- Cappetta, D.; Esposito, G.; Coppini, R.; Piegari, E.; Russo, R.; Ciuffreda, L.P.; Rivellino, A.; Santini, L.; Rafaniello, C.; Scavone, C.; et al. Effects of Ranolazine in a Model of Doxorubicin-induced Left Ventricle Diastolic Dysfunction. Br. J. Pharmacol. 2017, 174, 3696–3712. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial Calcium Overload Is a Key Determinant in Heart Failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Shi, Q.; Gao, S.; Song, L.; Zhao, Y.; Li, X.; Wu, J.L.; He, C.; Li, H.; Haifeng, Z. Comparative Proteomics Analysis of Differential Proteins in Respond to Doxorubicin Resistance in Myelogenous Leukemia Cell Lines. Proteome Sci. 2015, 13, 1. [Google Scholar] [CrossRef]
- Li, L.; Ni, J.; Li, M.; Chen, J.; Han, L.; Zhu, Y.; Kong, D.; Mao, J.; Wáng, Y.; Zhang, B.; et al. Ginsenoside Rg3 Micelles Mitigate Doxorubicin-Induced Cardiotoxicity and Enhance Its Anticancer Efficacy. Drug Deliv. 2017, 24, 1617–1630. [Google Scholar] [CrossRef]
- Starnes, L.M.; Hall, A.P.; Etal, D.; Cavallo, A.-L.; Grabowski, P.; Gallon, J.; Kha, M.; Hicks, R.; Pointon, A. RYR2 Deficient Human Model Identifies Calcium Handling and Metabolic Dysfunction Impacting Pharmacological Responses. Front. Cardiovasc. Med. 2024, 11, 1357315. [Google Scholar] [CrossRef]
- Joseph, L.C.; Reyes, M.V.; Homan, E.A.; Gowen, B.H.; Avula, U.M.R.; Goulbourne, C.N.; Wan, E.Y.; Elrod, J.W.; Morrow, J. The Mitochondrial Calcium Uniporter Promotes Arrhythmias Caused by High-Fat Diet. Sci. Rep. 2021, 11, 17808. [Google Scholar] [CrossRef]
- Genovese, I.; Fiorillo, A.; Ilari, A.; Masciarelli, S.; Fazi, F.; Colotti, G. Binding of Doxorubicin to Sorcin Impairs Cell Death and Increases Drug Resistance in Cancer Cells. Cell Death Dis. 2017, 8, e2950. [Google Scholar] [CrossRef]
- Zhang, N.; Ye, F.; Zhou, Y.; Zhu, W.; Xie, C.; Zheng, H.; Chen, H.; Chen, J.; Xie, X. Cardiac Ankyrin Repeat Protein Contributes to Dilated Cardiomyopathy and Heart Failure. FASEB J. 2021, 35, e21488. [Google Scholar] [CrossRef]
- Ramadan, M.; Sherman, M.; Jaimes, R.; Chaluvadi, A.; Swift, L.; Posnack, N.G. Disruption of Neonatal Cardiomyocyte Physiology Following Exposure to Bisphenol-A. Sci. Rep. 2018, 8, 7356. [Google Scholar] [CrossRef]
- Patintingan, C.G.H.; Louisa, M.; Juniantito, V.; Arozal, W.; Hanifah, S.; Wanandi, S.I.; Thandavarayan, R.A. Moringa Oleifera Leaves Extract Ameliorates Doxorubicin-Induced Cardiotoxicity via Its Mitochondrial Biogenesis Modulatory Activity in Rats. J. Exp. Pharmacol. 2023, 15, 307–319. [Google Scholar] [CrossRef]
- Li, Y.; Fan, L.; Wang, X.; Lv, S. Shenmai Injection Ameliorates Doxorubicin-Induced Myocardial Injury by Suppressing Autophagy-Apoptosis via miR-30a. Aging 2023, 15, 12400–12412. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.-K.; Jun, Y.; Dong, J.; Zhang, P.; Wan, L.; Li, K. MicroRNA-143 Increases Oxidative Stress and Myocardial Cell Apoptosis in a Mouse Model of Doxorubicin-Induced Cardiac Toxicity. Med. Sci. Monit. 2020, 26, e920394-1–e920394-12. [Google Scholar] [CrossRef]
- Karwt, R.; Bondar, O.V.; Pugachev, M.V.; Mohammad, T.; Kadyrova, A.S.; Pavelyev, R.S.; Alrhmoun, S.; Gnezdilov, O.I.; Shtyrlin, Y.G. Anticancer Potential of Pyridoxine-Based Doxorubicin Derivatives: An in Vitro Study. Life 2024, 14, 282. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Y.; Liu, N.; Liu, B. AP39, a Novel Mitochondria-Targeted Hydrogen Sulfide Donor Ameliorates Doxorubicin-Induced Cardiotoxicity by Regulating the AMPK/UCP2 Pathway. PLoS ONE 2024, 19, e0300261. [Google Scholar] [CrossRef]
- Poh, H.; Chiou, Y.; Chong, Q.Y.; Chen, R.-M.; Rangappa, K.S.; Ma, L.; Zhu, T.; Kumar, A.P.; Pandey, V.; Basappa, B.; et al. Inhibition of TFF3 Enhances Sensitivity—And Overcomes Acquired Resistance—To Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma. Cancers 2019, 11, 1528. [Google Scholar] [CrossRef]
- Yang, G.; Song, M.; Hoang, D.H.; Tran, Q.H.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. Melatonin Prevents Doxorubicin-Induced Cardiotoxicity Through Suppression of AMPKα2-dependent Mitochondrial Damage. Exp. Mol. Med. 2020, 52, 2055–2068. [Google Scholar] [CrossRef]
- Mohamed, D.S.; Shaban, N.S.; Labib, M.M.; Shehata, O. Ameliorative Effect of Almond Oil Against Doxorubicin-Induced Cardiotoxicity in Mice via Downregulation of TLR4 Gene Expression, Lowering NF-κB and TNF-α Levels. Adv. Anim. Vet. Sci. 2021, 10, 685–693. [Google Scholar] [CrossRef]
- Kanno, K.; Shimizu, K.; Shinoda, M.; Hayashi, M.; Takeichi, O.; Iwata, K. Role of Macrophage-Mediated Toll-Like Receptor 4–interleukin-1r Signaling in Ectopic Tongue Pain Associated With Tooth Pulp Inflammation. J. Neuroinflamm. 2020, 17, 312. [Google Scholar] [CrossRef]
- Zhang, F.; Dong, H.; Lv, T.; Jin, K.; Jin, Y.; Zhang, X.; Jiang, J. Moderate Hypothermia Inhibits Microglial Activation After Traumatic Brain Injury by Modulating Autophagy/Apoptosis and the MyD88-dependent TLR4 Signaling Pathway. J. Neuroinflamm. 2018, 15, 273. [Google Scholar] [CrossRef]
- Zhao, X.P.; Duan, L.; Zhao, Q.R.; Lv, X.; Tian, N.Y.; Yang, S.L.; Dong, K. NLRP3 inflammasome as a therapeutic target in doxorubicin-induced cardiotoxicity: Role of phytochemicals. Front. Pharmacol. 2025, 16, 1567312. [Google Scholar] [CrossRef]
- Knoops, B.; Becker, S.; Poncin, M.A.; Glibert, J.; Derclaye, S.; Clippe, A.; Alsteens, D. Specific Interactions Measured by AFM on Living Cells Between Peroxiredoxin-5 and TLR4: Relevance for Mechanisms of Innate Immunity. Cell Chem. Biol. 2018, 25, 550–559.e3. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.R.; Yin, H.; Wang, X. Small-Molecule Modulators of Toll-Like Receptors. Acc. Chem. Res. 2020, 53, 1046–1055. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Y.; Chen, R.; Wang, Y.; Fang, Y.; Qin, C.; Wang, T.; Shen, X.; Zhou, T.; Tian, L.; et al. Exosomal transfer of pro-pyroptotic miR-216a-5p exacerbates anthracycline cardiotoxicity through breast cancer-heart pathological crosstalk. Signal Transduct. Target. Ther. 2025, 10, 157. [Google Scholar] [CrossRef]
- Liu, A.; Bai, P.; You, H.; Zhuang, Z.; Tian, F.; Weng, H.; Wei, X.; Tang, L.; Wang, L.; Liu, C.; et al. SLAMF7 Restrains Pro-Inflammatory Macrophage Activation to Counteract Doxorubicin-Induced Cardiotoxicity. JACC Basic Transl. Sci. 2025. [Google Scholar] [CrossRef]
- Gambardella, J.; Santulli, G.; Fiordelisi, A.; Cerasuolo, F.A.; Wang, X.; Prevete, N.; Sommella, E.; Avvisato, R.; Buonaiuto, A.; Altobelli, G.G.; et al. Infiltrating macrophages amplify doxorubicin-induced cardiac damage: Role of catecholamines. Cell. Mol. Life Sci. 2023, 80, 323. [Google Scholar] [CrossRef]
- Tan, X.; Yan, C.; Zou, G.; Jing, R. Neurogenic differentiation 2 promotes inflammatory activation of macrophages in doxorubicin-induced myocarditis via regulating protein kinase D. BMC Cardiovasc. Disord. 2025, 25, 195. [Google Scholar] [CrossRef]
- Li, X.; Guo, D.; Chen, Y.; Hu, Y. Toll-Like Receptors/TNF-α Pathway Crosstalk and Impact on Different Sites of Recurrent Myocardial Infarction in Elderly Patients. Biomed. Res. Int. 2022, 2022, 1280350. [Google Scholar] [CrossRef]
- Zheng, X.; Li, S.; Yang, H. Roles of Toll-Like Receptor 3 in Human Tumors. Front. Immunol. 2021, 12, 667454. [Google Scholar] [CrossRef]
- Jegal, M.-E.; Jung, S.Y.; Han, Y.-S.; Kim, Y.-J. C-Terminal Truncated HBx Reduces Doxorubicin Cytotoxicity via ABCB1 Upregulation in Huh-7 Hepatocellular Carcinoma Cells. BMB Rep. 2019, 52, 330–335. [Google Scholar] [CrossRef]
- Al-malky, H.S.; Harthi, S.E.A.; Osman, A.M.M. Major Obstacles to Doxorubicin Therapy: Cardiotoxicity and Drug Resistance. J. Oncol. Pharm. Pract. 2019, 26, 434–444. [Google Scholar] [CrossRef]
- Li, L.; Zhong, J.; Li, M.; Sun, Y.; Niu, Y.; Wu, C.; Zhou, J.; Norton, N.; Li, Z.; Shi, Y.; et al. Disruption of MAP7D1 Gene Function Increases the Risk of Doxorubicin-Induced Cardiomyopathy and Heart Failure. Biomed. Res. Int. 2021, 2021, 8569921. [Google Scholar] [CrossRef]
- Yu, H.; Li, X.; Li, J.; He, Q.; Huang, M.; Tang, Y.; Chen, X.; Chen, J.; Tang, K.; Wei, C. H3K27ac Acts as a Molecular Switch for Doxorubicin-Induced Activation of Cardiotoxic Genes. Clin. Epigenetics 2024, 16, 91. [Google Scholar] [CrossRef]
- Mendonca, A.; Sánchez, O.F.; Zhao, H.; Lin, L.; Min, A.; Yuan, C. Development and Application of Novel BiFC Probes for Cell Sorting Based on Epigenetic Modification. Cytom. Part A 2022, 101, 339–350. [Google Scholar] [CrossRef]
- Koh, J.Y.P.; Itahana, Y.; Mendenhall, I.H.; Low, D.H.W.; Soh, E.X.Y.; Guo, A.K.; Chionh, Y.T.; Wang, L.F.; Itahana, K. ABCB1 Protects Bat Cells From DNA Damage Induced by Genotoxic Compounds. Nat. Commun. 2019, 10, 2820. [Google Scholar] [CrossRef]
- Poma, P.; Rigogliuso, S.; Labbozzetta, M.; Nicosia, A.; Costa, S.; Ragusa, M.A.; Notarbartolo, M. Epigenetic and Cellular Reprogramming of Doxorubicin-Resistant McF-7 Cells Treated With Curcumin. Int. J. Mol. Sci. 2024, 25, 13416. [Google Scholar] [CrossRef]
- Robinson, E.; Ameri, P.; Delrue, L.; Vanderheyden, M.; Bartúnek, J.; Altieri, P.; Heymans, S.; Heggermont, W. Differential Expression of Epigenetic Modifiers in Early and Late Cardiotoxic Heart Failure Reveals DNA Methylation as a Key Regulator of Cardiotoxicity. Front. Cardiovasc. Med. 2023, 10, 884174. [Google Scholar] [CrossRef]
- Gao, F.; Xu, T.; Zang, F.; Luo, Y.; Pan, D. Cardiotoxicity of Anticancer Drugs: Molecular Mechanisms, Clinical Management and Innovative Treatment. Drug Des. Dev. Ther. 2024, 18, 4089–4116. [Google Scholar] [CrossRef]
- Sarno, F.; Benincasa, G.; List, M.; Barabási, A.-L.; Baumbach, J.; Ciardiello, F.; Filetti, S.; Glass, K.; Loscalzo, J.; Marchese, C.; et al. Clinical Epigenetics Settings for Cancer and Cardiovascular Diseases: Real-Life Applications of Network Medicine at the Bedside. Clin. Epigenetics 2021, 13, 66. [Google Scholar] [CrossRef]
- Hagag, A.A.; Badraia, I.M.; El-Shehaby, W.A.; Mabrouk, M.M. Protective Role of Black Seed Oil in Doxorubicin-Induced Cardiac Toxicity in Children With Acute Lymphoblastic Leukemia. J. Oncol. Pharm. Pract. 2020, 26, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Agostinucci, K.; Grant, M.; Seelig, D.; Yücel, D.; Berlo, J.H.v.; Bartolomucci, A.; Dyck, J.R.; Zordoky, B.N. Divergent Cardiac Effects of Angiotensin II and Isoproterenol Following Juvenile Exposure to Doxorubicin. Front. Cardiovasc. Med. 2022, 9, 742193. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Singh, P.; Cejas, R.B.; Zhou, L.; Sharafeldin, N.; Trainor, P.J.; Landier, W.; Cheng, C.; Hageman, L.; Wang, F.; et al. DNA Damage Response and Repair Genes and Anthracycline-Induced Cardiomyopathy in Childhood Cancer Survivors: A Report From the Children’s Oncology Group and the Childhood Cancer Survivor Study. Circ. Genom. Precis. Med. 2025, 18, e004813. [Google Scholar] [CrossRef]
- Camilli, M.; Cipolla, C.M.; Dent, S.; Minotti, G.; Cardinale, D.M. Anthracycline Cardiotoxicity in Adult Cancer Patients: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2024, 6, 655–677. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cheng, C.; Fan, L.; Xu, X.; Chen, J.; Yang, F.; Tang, Y.; Yang, C. Assessment of Left Heart Dysfunction to Predict Doxorubicin Cardiotoxicity in Children With Lymphoma. Front. Pediatr. 2023, 11, 1163664. [Google Scholar] [CrossRef]
- Cheah, I.K.; Tang, R.M.; Wang, X.; Sachaphibulkij, K.; Chong, S.Y.; Lim, L.H.K.; Wang, J.W.; Halliwell, B. Protection Against Doxorubicin-Induced Cardiotoxicity by Ergothioneine. Antioxidants 2022, 12, 320. [Google Scholar] [CrossRef]
- Wan, G.; Ji, L.; Xia, W.; Cheng, L.; Zhang, Y. Bioinformatics Identification of Potential Candidate Blood Indicators for Doxorubicin-induced Heart Failure. Exp. Ther. Med. 2018, 16, 2534–2544. [Google Scholar] [CrossRef]
- Kopp, L.M.; Womer, R.B.; Schwartz, C.L.; Ebb, D.H.; Franco, V.I.; Hall, D.; Barkauskas, D.A.; Krailo, M.; Grier, H.E.; Meyers, P.A.; et al. Effects of Dexrazoxane on Doxorubicin-Related Cardiotoxicity and Second Malignant Neoplasms in Children With Osteosarcoma: A Report From the Children’s Oncology Group. Cardio-Oncology 2019, 5, 15. [Google Scholar] [CrossRef]
- Curren, V.; Dham, N.; Spurney, C.F. Diagnosis, Prevention, Treatment and Surveillance of Anthracycline-Induced Cardiovascular Toxicity in Pediatric Cancer Survivors. Hearts 2021, 2, 45–60. [Google Scholar] [CrossRef]
- Bertorello, N.; Luksch, R.; Bisogno, G.; Haupt, R.; Spallarossa, P.; Cenna, R.; Fagioli, F. Cardiotoxicity in Children With Cancer Treated With Anthracyclines: A Position Statement on Dexrazoxane. Pediatr. Blood Cancer 2023, 70, e30515. [Google Scholar] [CrossRef]
- Reichardt, P.; Tabone, M.D.; Mora, J.; Morland, B.; Jones, R.L. Risk-benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: Re-evaluating the European labeling. Future Oncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef]
- Carfagnini, C.; Bechara, S.; Kandula, M. The Risk of Peripartum Cardiomyopathy Among Pediatric, Adolescent, and Young Adult Cancer Patients Exposed to Doxorubicin: An Opinion Article. Front. Oncol. 2024, 14, 1395465. [Google Scholar] [CrossRef]
- Perez, I.E.; Alam, S.T.; Hernandez, G.A.; Sancassani, R. Cancer Therapy-Related Cardiac Dysfunction: An Overview for the Clinician. Clin. Med. Insights Cardiol. 2019, 13, 13. [Google Scholar] [CrossRef]
- Quagliariello, V.; Di Mauro, A.; Ferrara, G.; Bruzzese, F.; Palma, G.; Luciano, A.; Canale, M.L.; Bisceglia, I.; Iovine, M.; Cadeddu Dessalvi, C.; et al. Cardio-Renal and Systemic Effects of SGLT2i Dapagliflozin on Short-Term Anthracycline and HER-2-Blocking Agent Therapy-Induced Cardiotoxicity. Antioxidants 2025, 14, 612. [Google Scholar] [CrossRef]
- Cronin, M.J.; Seher, M.; Arsang-Jang, S.; Lowery, A.; Kerin, M.J.; Wijns, W.; Soliman, O. Multimodal Imaging of Cancer Therapy-Related Cardiac Dysfunction in Breast Cancer—A State-of-the-Art Review. J. Clin. Med. 2023, 12, 6295. [Google Scholar] [CrossRef]
- Wan, Y.; He, B.; Zhu, D.; Wang, L.; Huang, R.; Wang, S.; Wang, C.H.; Zhang, M.; Ma, L.; Gao, F. Nicorandil Ameliorates Doxorubicin-Induced Cardiotoxicity in Rats, as Evaluated by 7 T Cardiovascular Magnetic Resonance Imaging. Cardiovasc. Drugs Ther. 2021, 37, 39–51. [Google Scholar] [CrossRef]
- Azcona, J.A.; Wacker, A.; Lee, C.; Fung, E.K.; Jeitner, T.M.; Manzo, O.L.; Lorenzo, A.D.; Babich, J.W.; Amor-Coarasa, A.; Kelly, J.M. 2-[18F]Fluoropropionic Acid PET Imaging of Doxorubicin-Induced Cardiotoxicity. Mol. Imaging Biol. 2024, 27, 109–119. [Google Scholar] [CrossRef]
- Cochera, F.; Dinca, D.; Bordejevic, D.A.; Citu, I.M.; Mavrea, A.; Andor, M.; Trofenciuc, N.-M.; Tomescu, M. Nebivolol Effect on Doxorubicin-Induced Cardiotoxicity in Breast Cancer. Cancer Manag. Res. 2018, 10, 2071–2081. [Google Scholar] [CrossRef]
- Huang, C.; Zhu, S. Doxorubicin-Induced Cardiomyopathy: Mechanisms, Diagnosis and Therapeutic Drugs. Highlights Sci. Eng. Technol. 2022, 6, 129–135. [Google Scholar] [CrossRef]
- Michel, L.; Rassaf, T.; Totzeck, M. Biomarkers for the Detection of Apparent and Subclinical Cancer Therapy-Related Cardiotoxicity. J. Thorac. Dis. 2018, 10, S4282–S4295. [Google Scholar] [CrossRef] [PubMed]
- Lambrinou, E.; DeCourcey, J.; Hill, L. Personalizing Heart Failure Care to the Patient With Cancer. Curr. Heart Fail. Rep. 2022, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Frères, P.; Bouznad, N.; Servais, L.; Josse, C.; Wenric, S.; Poncin, A.; Thiry, J.; Moonen, M.; Oury, C.; Lancellotti, P.; et al. Variations of Circulating Cardiac Biomarkers During and After Anthracycline-Containing Chemotherapy in Breast Cancer Patients. BMC Cancer 2018, 18, 102. [Google Scholar] [CrossRef]
- Iacobescu, L.; Ciobanu, A.; Corlatescu, A.-D.; Simionescu, M.; Iacobescu, G.L.; Dragomir, E.; Vinereanu, D. The Role of Circulating MicroRNAs in Cardiovascular Diseases: A Novel Biomarker for Diagnosis and Potential Therapeutic Targets? Cureus 2024, 16, e64100. [Google Scholar] [CrossRef]
- Mansouri, F.; Mohammadzad, M.H.S. Up-Regulation of Cell-Free MicroRNA-1 and MicroRNA-221-3p Levels in Patients With Myocardial Infarction Undergoing Coronary Angiography. Adv. Pharm. Bull. 2020, 11, 719–727. [Google Scholar] [CrossRef]
- Poovorawan, N.; Susiriwatananont, T.; Teerapakpinyo, C.; Chariyavilaskul, P.; Sitthideatphaiboon, P.; Jarutasnangkul, L.; Tumkosit, M.; Chattranukulchai, P.; Theerasuwipakorn, N.; Aporntewan, C.; et al. Long-term impact of anthracycline in early-stage breast cancer, bridging of MiRNAs profiler for early cardiotoxicity. Cardiooncology 2025, 11, 39. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Platt, C.; Hastings, M.H.; Chen, W.C.W.; Guerra, J.R.B.; Tokuyama, T.; Torizal, F.G.; Liu, W.; Obana, T.; Bayer, A.L.; et al. Mitigation of Doxorubicin Cardiotoxicity With Synergistic miRNA Combinations Identified Using Combinatorial Genetics en masse (CombiGEM). JACC CardioOncol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Klotzka, A.; Iwańczyk, S.; Ropacka-Lesiak, M.; Misan, N.; Lesiak, M. Anthracycline-Induced Microcirculation Disorders: AIM PILOT Study. Kardiol. Pol. 2023, 81, 766–768. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Kassem, H.H. Protective Effect of Nebivolol on Doxorubicin-Induced Cardiotoxicity in Rats. Arch. Med. Sci. 2018, 14, 1450–1458. [Google Scholar] [CrossRef]
- Han, S.; An, T.; Liu, W.; Song, Y.; Zhu, J. Secondary Lymphoma Develops in the Setting of Heart Failure When Treating Breast Cancer: A Case Report. World J. Clin. Cases 2019, 7, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; VanDongen, N.S.; D’Alessandro, A.; Reisz, J.A.; Ziemba, B.P.; Seals, D.R. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. Cardiooncology 2020, 2, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Galán-Arriola, C.; Vilchez-Tschischke, J.P.; Lobo, M.; López, G.J.; Molina, A.; Pérez-Martínez, C.; Villena-Gutiérrez, R.; Macías, Á.; Díaz-Rengifo, I.A.; Oliver, E.; et al. Coronary Microcirculation Damage in Anthracycline Cardiotoxicity. Cardiovasc. Res. 2021, 118, 531–541. [Google Scholar] [CrossRef]
- Abdelgawad, I.Y.; Agostinucci, K.; Ismail, S.G.; Grant, M.; Zordoky, B.N. EA.hy926 Cells and HUVECs Share Similar Senescence Phenotypes but Respond Differently to the Senolytic Drug ABT-263. Cells 2022, 11, 1992. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Kakeshita, K.; Imamura, T.; Shima, T.; Fujioka, H.; Yamazaki, H.; Koike, T.; Kinugawa, K. Pegylated-Liposomal Doxorubicin-Induced Glomerular Thrombotic Microangiopathy. Intern. Med. 2024, 63, 2839–2845. [Google Scholar] [CrossRef]
- Venturini, W.; Olate-Briones, A.; Valenzuela, C.; Méndez, D.; Fuentes, E.; Cayo, Á.; Mancilla, D.; Segovia, R.; Brown, N.E.; Moore-Carrasco, R. Platelet Activation Is Triggered by Factors Secreted by Senescent Endothelial HMEC-1 Cells in Vitro. Int. J. Mol. Sci. 2020, 21, 3287. [Google Scholar] [CrossRef]
- Xiang, G.; Shi, T.; Nwaele, C.O.; Xiao, H.; Liu, Y.; Wang, Q.; Zhang, J.; Zheng, Y. Inhibition of the Sp1/PI3K/AKT signaling pathway exacerbates doxorubicin-induced cardiomyopathy. Biochim. Biophys. Acta Mol. Cell Res. 2025, 1872, 119960. [Google Scholar] [CrossRef]
- Chen, L.; Holder, R.; Porter, C.; Shah, Z. Vitamin D3 Attenuates Doxorubicin-Induced Senescence of Human Aortic Endothelial Cells by Upregulation of IL-10 via the pAMPKα/Sirt1/Foxo3a Signaling Pathway. PLoS ONE 2021, 16, e0252816. [Google Scholar] [CrossRef]
- Li, C.; Cheung, M.K.H.; Han, S.; Zhang, Z.; Chen, L.; Chen, J.; Zeng, H.; Qiu, J. Mesenchymal Stem Cells and Their Mitochondrial Transfer: A Double-Edged Sword. Biosci. Rep. 2019, 39, BSR20182417. [Google Scholar] [CrossRef]
- Wolle, C.F.B.; Zollmann, L.d.A.; Etges, A.; Vitalis, G.S.; Leite, C.E.; Campos, M.M. Effects of the Antioxidant Agent Tempol on Periapical Lesions in Rats With Doxorubicin-Induced Cardiomyopathy. J. Endod. 2012, 38, 191–195. [Google Scholar] [CrossRef]
- Podyacheva, E.; Шмaкoвa, T.B.; Кушнaревa, E.A.; Onopchenko, A.; Martynov, M.; Andreeva, D.; Toropov, R.I.; Cheburkin, Y.V.; Levchuk, K.; Goldaeva, A.; et al. Modeling Doxorubicin-Induced Cardiomyopathy With Fibrotic Myocardial Damage in Wistar Rats. Cardiol. Res. 2022, 13, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, A.; Guo, C.; Chun-zhi, S.; Zhang, Y.; Liu, Q.; Sparatore, A.; Wang, C. S-Diclofenac Protects Against Doxorubicin-Induced Cardiomyopathy in Mice via Ameliorating Cardiac Gap Junction Remodeling. PLoS ONE 2011, 6, e26441. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Singhal, T.; Sharma, H. Cardioprotective Effect of Ammonium Glycyrrhizinate Against Doxorubicin-Induced Cardiomyopathy in Experimental Animals. Indian J. Pharmacol. 2014, 46, 527. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.I.; Saba, S.; Ammar, R.I.; El-Sayed, L.; Ghaly, W.; Dhingra, S. Erythropoietin Protects Against Doxorubicin-Induced Heart Failure. AJP Heart Circ. Physiol. 2011, 301, H2413–H2421. [Google Scholar] [CrossRef]
- Wang, F.; Han, L. Upregulation of Serum and Glucocorticoid-Regulated Kinase 1 (SGK1) Ameliorates Doxorubicin-Induced Cardiotoxic Injury, Apoptosis, Inflammation and Oxidative Stress by Suppressing Glucose Regulated Protein 78 (GRP78)-mediated Endoplasmic Reticulum Stress. Bioengineered 2021, 13, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lan, R.; Guo, Z.; Cai, S.; Wang, J.; Wang, Q.; Li, Z.; Li, Z.; Wang, Q.; Li, J.; et al. Histone Demethylase JMJD3 Mediated Doxorubicin-Induced Cardiomyopathy by Suppressing SESN2 Expression. Front. Cell Dev. Biol. 2020, 8, 548605. [Google Scholar] [CrossRef]
- Liu, Y.; Asnani, A.; Zou, L.; Bentley, V.L.; Yu, M.; Wang, Y.; Dellaire, G.; Sarkar, K.S.; Dai, M.; Chen, H.H.; et al. Visnagin Protects Against Doxorubicin-Induced Cardiomyopathy Through Modulation of Mitochondrial Malate Dehydrogenase. Sci. Transl. Med. 2014, 6, 266ra170. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Wang, S.; He, X.; Liu, M.; Bai, B.; Tian, C.; Sun, R.; Yu, T.; Chu, X.M. Role of Acetylation in Doxorubicin-Induced Cardiotoxicity. Redox Biol. 2021, 46, 102089. [Google Scholar] [CrossRef]
- Dong, Q.; Chen, L.; Lu, Q.; Sharma, S.; Li, L.; Morimoto, S.; Wang, G. Quercetin Attenuates Doxorubicin Cardiotoxicity by Modulating Bmi-1 Expression. Br. J. Pharmacol. 2014, 171, 4440–4454. [Google Scholar] [CrossRef]
- Ma, X.; Ding, Y.; Wang, Y.; Xu, X. A Doxorubicin-Induced Cardiomyopathy Model in Adult Zebrafish. J. Vis. Exp. 2018, 136, 57567. [Google Scholar] [CrossRef]
- Yerebakan, C.; Boltze, J.; Elmontaser, H.; Yörüker, U.; Latus, H.; Khalil, M.; Ostermayer, S.; Steinbrenner, B.; Apitz, C.; Schneider, M.; et al. Effects of Pulmonary Artery Banding in Doxorubicin-Induced Left Ventricular Cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2019, 157, 2416–2428.e2414. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Lukovic, D.; Zlabinger, K.; Spannbauer, A.; Gugerell, A.; Pavo, N.; Traxler, D.; Pils, D.; Maurer, G.; Jakab, A.; et al. Liposomal Doxorubicin Attenuates Cardiotoxicity via Induction of Interferon-Related DNA Damage Resistance. Cardiovasc. Res. 2020, 116, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Medina-Hernandez, D.; Cadiz, L.; Mastrangelo, A.; Moreno-Arciniegas, A.; Fernandez Tocino, M.; Cueto Becerra, A.A.; Diaz-Guerra Priego, A.; Skoza, W.A.; Higuero-Verdejo, M.I.; Lopez-Martin, G.J.; et al. SGLT2i Therapy Prevents Anthracycline-Induced Cardiotoxicity in a Large Animal Model by Preserving Myocardial Energetics. JACC CardioOncol. 2025, 7, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Kucukseymen, S.; Cai, X.; Yankama, T.; Rodriguez, J.; Sai, E.; Pierce, P.; Ngo, L.; Nakamori, S.; Tung, N.; et al. Cardiovascular magnetic resonance characterization of myocardial tissue injury in a miniature swine model of cancer therapy-related cardiovascular toxicity. J. Cardiovasc. Magn. Reson. 2024, 26, 101033. [Google Scholar] [CrossRef]
- Chang, H.M.; Hsu, J.Y.; Ahn, C.; Yeh, E.T.H. Prevention of Heart Failure Induced by Doxorubicin with Early Administration of Dexrazoxane (PHOENIX Study): Dose response and time course of dexrazoxane-induced degradation of topoisomerase 2b. Cardiooncology 2025, 11, 42. [Google Scholar] [CrossRef]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef]
- Walker, D.M.; Fisher, B.T.; Seif, A.E.; Huang, Y.S.; Torp, K.; Li, Y.; Aplenc, R. Dexrazoxane use in pediatric patients with acute lymphoblastic or myeloid leukemia from 1999 and 2009: Analysis of a national cohort of patients in the Pediatric Health Information Systems database. Pediatr. Blood Cancer 2013, 60, 616–620. [Google Scholar] [CrossRef]
- Henriksen, P.A.; Hall, P.; MacPherson, I.R.; Joshi, S.S.; Singh, T.; Maclean, M.; Lewis, S.; Rodriguez, A.; Fletcher, A.; Everett, R.J.; et al. Multicenter, Prospective, Randomized Controlled Trial of High-Sensitivity Cardiac Troponin I-Guided Combination Angiotensin Receptor Blockade and Beta-Blocker Therapy to Prevent Anthracycline Cardiotoxicity: The Cardiac CARE Trial. Circulation 2023, 148, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.; Maier, R.H.; Akhter, N.; Sayari, M.; Ogundimu, E.; Maddox, J.M.; Vahabi, S.; Humphreys, A.C.; Graham, J.; Oxenham, H.; et al. Preventing Cardiac Damage in Patients Treated for Breast Cancer and Lymphoma: The PROACT Clinical Trial. JACC CardioOncol. 2024, 6, 684–696. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Moustafa, I.; Viljoen, M.; Perumal-Pillay, V.A.; Oosthuizen, F. Critical Appraisal of Clinical Guidelines for Prevention and Management of Doxorubicin-Induced Cardiotoxicity. J. Oncol. Pharm. Pract. 2023, 29, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hao, C.; Ji, Z.; Qu, Y.; Zuo, W.; Yang, M.; Zuo, P.; Carvalho, A.; Ma, G.; Li, Y. Upregulation of Biomarker Limd1 Was Correlated With Immune Infiltration in Doxorubicin-Related Cardiotoxicity. Mediat. Inflamm. 2023, 2023, 8347759. [Google Scholar] [CrossRef] [PubMed]
- Heemelaar, J.C.; Janson, J.A.; Braun, J.; Speetjens, F.M.; Michiel, A.J.v.d.S.; Hugo, J.d.V.; Barge-Schaapveld, D.Q.; Beeres, S.L.; Tops, L.F.; Gelderblom, H.; et al. Case Report: Challenges in Monitoring and Treatment of Anthracycline Induced Cardiotoxicity in Young Adults With Osteosarcoma. Cardio-Oncology 2022, 8, 18. [Google Scholar] [CrossRef]
- Gunsaulus, M.; Alsaied, T.; Tersak, J.M.; Friehling, E.; Rose-Felker, K. Abnormal Global Longitudinal Strain During Anthracycline Treatment Predicts Future Cardiotoxicity in Children. Pediatr. Cardiol. 2023, 45, 1750–1758. [Google Scholar] [CrossRef]
- Nakayama, T.; Oshima, Y.; Kusumoto, S.; Yamamoto, J.; Osaga, S.; Fujinami, H.; Kikuchi, T.; Suzuki, T.; Totani, H.; Kinoshita, S.; et al. Clinical Features of Anthracycline-induced Cardiotoxicity in Patients With Malignant Lymphoma Who Received a CHOP Regimen With or Without Rituximab: A Single-center, Retrospective Observational Study. eJHaem 2020, 1, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, R.B.; Aljizeeri, A.; Alsaileek, A.; Alrashid, A.; Alolayan, A.; Alkaiyat, M.; Alenazy, B.; Shehata, H.; Alqahtani, J.; Ardah, H.I.; et al. Cardiac Morbidity and Mortality in Patients With Sarcoma: A Population-Based Study. Clin. Med. Insights Oncol. 2024, 18, 11795549241237703. [Google Scholar] [CrossRef]
- Denysova, M.V.; Strutynska, N.A.; Mys, L.A.; Korkach, Y.P.; Sagach, V.F.; Strutynskyi, R.B. Activation of ATP-sensitive Potassium Channels Prevents Doxorubicin-Induced Mitochondrial Dysfunction in the Heart and Impaired Vascular Responses in Rats. Fiziolohichnyĭ Zhurnal 2024, 70, 3–11. [Google Scholar] [CrossRef]
- Chen, W.; Kim, S.J.; Kim, S.; Beheshtian, C.; Kim, N.S.; Shin, K.-H.; Kim, R.H.; Kim, S.; Park, N.-H. GV1001, hTERT Peptide Fragment, Prevents Doxorubicin-Induced Endothelial-to-Mesenchymal Transition in Human Endothelial Cells and Atherosclerosis in Mice. Cells 2025, 14, 98. [Google Scholar] [CrossRef]
- Lv, H.; Tan, R.; Liao, J.; Hao, Z.; Yang, X.; Liu, Y.; Xia, Y. Doxorubicin Contributes to Thrombus Formation and Vascular Injury by Interfering With Platelet Function. Ajp Heart Circ. Physiol. 2020, 319, H133–H143. [Google Scholar] [CrossRef]
- Norton, N.; Crook, J.E.; Wang, L.; Olson, J.E.; Kachergus, J.M.; Serie, D.; Necela, B.M.; Borgman, P.G.; Advani, P.; Ray, J.; et al. Association of Genetic Variants at TRPC6 With Chemotherapy-Related Heart Failure. Front. Cardiovasc. Med. 2020, 7, 142. [Google Scholar] [CrossRef]
- Li, M.Y.; Peng, L.M.; Chen, X.P. Pharmacogenomics in drug-induced cardiotoxicity: Current status and the future. Front. Cardiovasc. Med. 2022, 9, 966261. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.; Wen Tan, W.L.; Rhee, J.W.; Wu, J.C. Modeling Susceptibility to Cardiotoxicity in Cancer Therapy Using Human iPSC-Derived Cardiac Cells and Systems Biology. Heart Fail. Clin. 2022, 18, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Raisi-Estabragh, Z.; Murphy, A.C.; Ramalingam, S.; Scherrer-Crosbie, M.; Lopez-Fernandez, T.; Reynolds, K.L.; Aznar, M.; Lin, A.E.; Libby, P.; Cordoba, R.; et al. Cardiovascular Considerations Before Cancer Therapy: Gaps in Evidence and JACC: CardioOncology Expert Panel Recommendations. JACC CardioOncol. 2024, 6, 631–654. [Google Scholar] [CrossRef]
- Huang, Y.; Song, C.; He, J.; Li, M. Research Progress in Endothelial Cell Injury and Repair. Front. Pharmacol. 2022, 13, 997272. [Google Scholar] [CrossRef]
- Yu, L.; Liang, Q.; Zhang, W.; Liao, M.; Wen, M.; Zhan, B.; Bao, H.; Cheng, X. HSP22 Suppresses Diabetes-Induced Endothelial Injury by Inhibiting Mitochondrial Reactive Oxygen Species Formation. Redox Biol. 2019, 21, 101095. [Google Scholar] [CrossRef] [PubMed]
- Sogawa, Y.; Nagasu, H.; Itano, S.; Kidokoro, K.; Taniguchi, S.; Takahashi, M.; Kadoya, H.; Satoh, M.; Sasaki, T.; Kashihara, N. The eNOS-NO Pathway Attenuates Kidney Dysfunction via Suppression of Inflammasome Activation in Aldosterone-Induced Renal Injury Model Mice. PLoS ONE 2018, 13, e0203823. [Google Scholar] [CrossRef]
- Liu, Y.; Honglin, Y.; Li, C.; Jiang, F.; Zhang, S.; Zhang, X.; Li, Y. Sinapine Thiocyanate Ameliorates Vascular Endothelial Dysfunction in Hypertension by Inhibiting Activation of the NLRP3 Inflammasome. Front. Pharmacol. 2021, 11, 620159. [Google Scholar] [CrossRef]
- Nie, M.; Lei, D.; Liu, Z.; Zeng, K.; Liang, X.; Huang, P.; Wang, Y.; Sun, P.; Yang, H.; Liu, P.; et al. Cardiomyocyte-Localized CCDC25 Senses NET DNA to Promote Doxorubicin Cardiotoxicity by Activating Autophagic Flux. Nat. Cancer, 2025; in press. [Google Scholar]
- Tillman, L.; Margalef Rieres, J.; Ahjem, E.; Bishop-Guest, F.; McGrath, M.; Hatrick, H.; Pranjol, M.Z.I. Thinking Outside the Therapeutic Box: The Potential of Polyphenols in Preventing Chemotherapy-Induced Endothelial Dysfunction. Cells 2025, 14, 566. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhutani, V.; Varzideh, F.; Wilson, S.; Kansakar, U.; Jankauskas, S.S.; Santulli, G. Doxorubicin-Induced Cardiotoxicity: A Comprehensive Update. J. Cardiovasc. Dev. Dis. 2025, 12, 207. https://doi.org/10.3390/jcdd12060207
Bhutani V, Varzideh F, Wilson S, Kansakar U, Jankauskas SS, Santulli G. Doxorubicin-Induced Cardiotoxicity: A Comprehensive Update. Journal of Cardiovascular Development and Disease. 2025; 12(6):207. https://doi.org/10.3390/jcdd12060207
Chicago/Turabian StyleBhutani, Vasvi, Fahimeh Varzideh, Scott Wilson, Urna Kansakar, Stanislovas S. Jankauskas, and Gaetano Santulli. 2025. "Doxorubicin-Induced Cardiotoxicity: A Comprehensive Update" Journal of Cardiovascular Development and Disease 12, no. 6: 207. https://doi.org/10.3390/jcdd12060207
APA StyleBhutani, V., Varzideh, F., Wilson, S., Kansakar, U., Jankauskas, S. S., & Santulli, G. (2025). Doxorubicin-Induced Cardiotoxicity: A Comprehensive Update. Journal of Cardiovascular Development and Disease, 12(6), 207. https://doi.org/10.3390/jcdd12060207