
Genetic Spectrum of Lithuanian Familial Hypercholesterolemia Patients


 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Materials and Methods
- Definite FH (DLCN score > 8)
- Probable FH (DLCN score 6–8)
- Possible FH (DLCN score 3–5)
- Unlikely FH (DLCN score < 3)
- Age from 0 to 17 years for children and from 18 to 85 for adults.
- Elevated LDL-C (>4.9 mmol/L in adults and >3.9 mmol/L in children).
- Clinical suspicion of FH.
- Availability of data for initial DLCN scoring in adult patients.
- Secondary causes of hyperlipidemia (e.g., untreated hypothyroidism, cholestasis, and nephrotic syndrome).
- End-stage oncological or somatic disease.
- Pregnancy.
- Clinically significant cerebrovascular disease.
- Inability to provide informed consent by themselves or their legal guardian.
3. Statistical Analysis
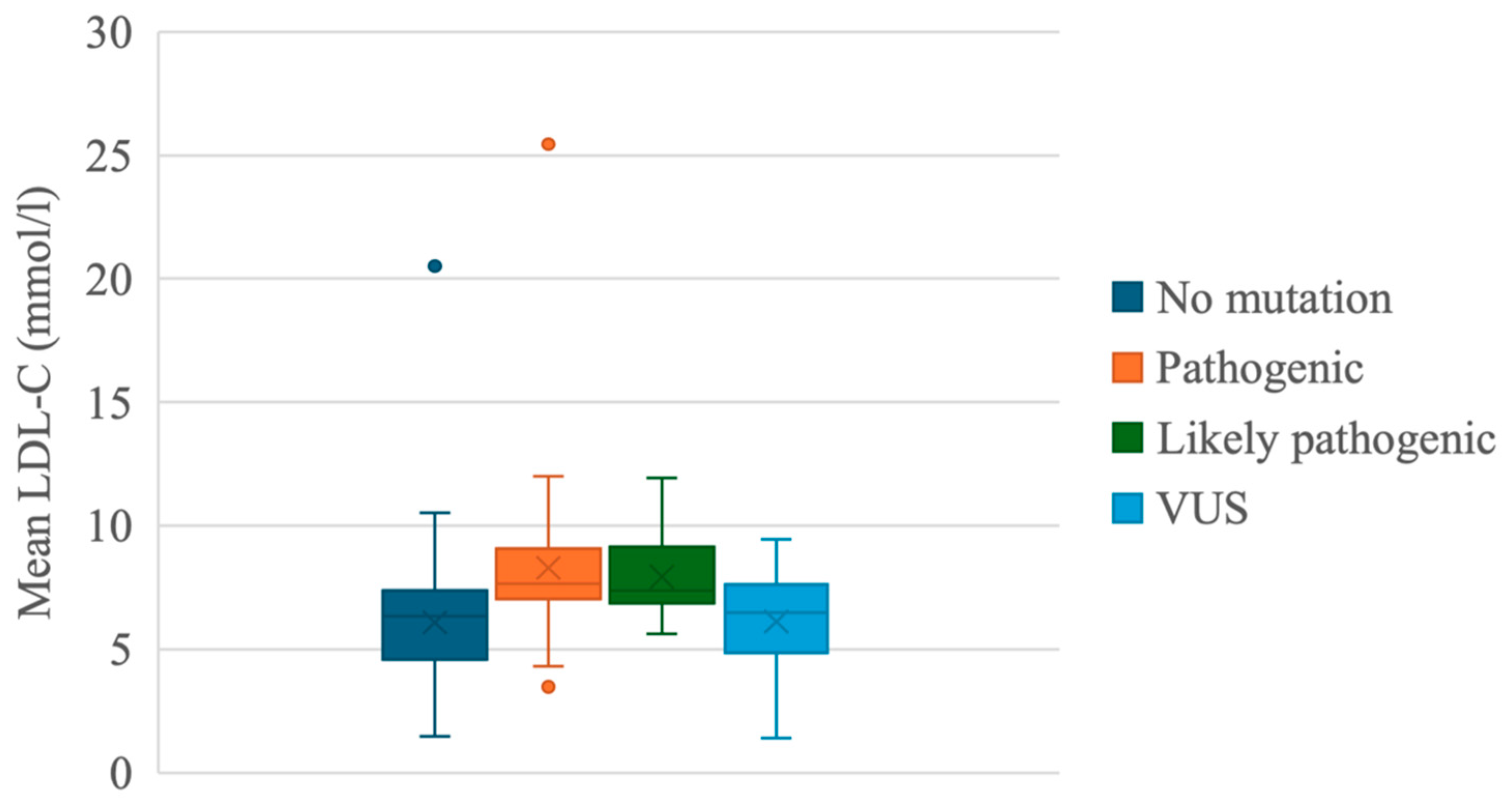
4. Results
5. Discussion
6. Conclusions
7. Limitations of the Study
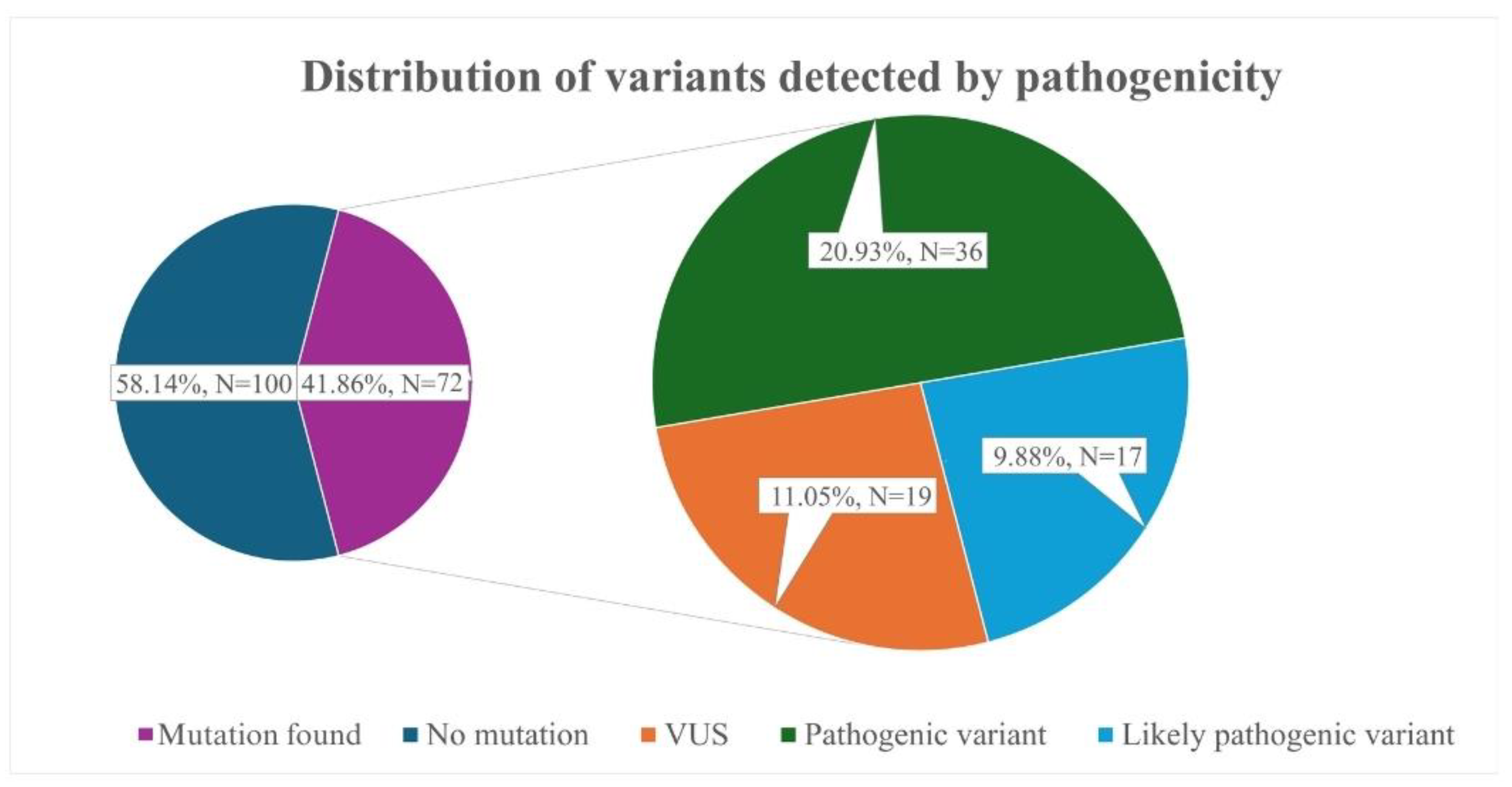
- Our study identified 36 pathogenic variants in the LDLR, APOB, and LDLRAP1 genes. Frequently identified variants were LDLR c.654_656del p.(Gly219del) and APOB c.10580G>A p.(Arg3527Gln), which are both associated with the founder’s effect.
- Genetic testing increased the number of patients classified as “FH” according to the Dutch Lipid Clinic Network criteria by nearly 86.2% in our cohort.
- Women in our cohort were diagnosed with FH nine years later than men, supporting claims of gender disparities in FH diagnostics.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diboun, I.; Al-Sarraj, Y.; Toor, S.M.; Mohammed, S.; Qureshi, N.; Al Hail, M.S.H.; Jayyousi, A.; Al Suwaidi, J.; Albagha, O.M.E. The Prevalence and Genetic Spectrum of Familial Hypercholesterolemia in Qatar Based on Whole Genome Sequencing of 14,000 Subjects. Front. Genet. 2022, 13, 927504. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef]
- van der Crabben, S.N.; Mörner, S.; Lundström, A.C.; Jonasson, J.; Bikker, H.; Amin, A.S.; Rydberg, A.; Wilde, A.A.M. Should variants of unknown significance (VUS) be disclosed to patients in cardiogenetics or not; only in case of high suspicion of pathogenicity? Eur. J. Hum. Genet. 2022, 30, 1208–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Facio, F.M.; Aradhya, K.W.; Rojahn, S.; Hatchell, K.E.; Aguilar, S.; Ouyang, K.; Saitta, S.; Hanson-Kwan, A.K.; Capurro, N.N.; et al. Rates and Classification of Variants of Uncertain Significance in Hereditary Disease Genetic Testing. JAMA Netw. Open 2023, 6, e2339571. [Google Scholar] [CrossRef]
- Pejic, R.N. Familial hypercholesterolemia. Ochsner J. 2014, 14, 669–672. [Google Scholar] [PubMed]
- Vaezi, Z.; Amini, A. Familial Hypercholesterolemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK556009/ (accessed on 9 March 2025).
- Kubilius, R. Kardiologijos Pagrindai: Universiteto Vadovėlis; Kauno Krašto Kardiologų Draugija: Kaunas, Lithuania, 2019; Available online: https://hdl.handle.net/20.500.12512/21971 (accessed on 9 March 2025).
- Abifadel, M.; Boileau, C. Genetic and molecular architecture of familial hypercholesterolemia. J. Intern. Med. 2023, 293, 144–165. [Google Scholar] [CrossRef]
- Meiner, V.; Landsberger, D.; Berkman, N.; Reshef, A.; Segal, P.; Seftel, H.C.; van der Westhuyzen, D.R.; Jeenah, M.S.; Coetzee, G.A.; Leitersdorf, E. A common Lithuanian mutation causing familial hypercholesterolemia in Ashkenazi Jews. Am. J. Hum. Genet. 1991, 49, 443–449. [Google Scholar] [PubMed]
- Nissen, P.H.; Damgaard, D.; Stenderup, A.; Nielsen, G.G.; Larsen, M.L.; Faergeman, O. Genomic characterization of five deletions in the LDL receptor gene in Danish Familial Hypercholesterolemic subjects. BMC Med. Genet. 2006, 7, 55. [Google Scholar] [CrossRef]
- Miyake, Y.; Tajima, S.; Funahashi, T.; Yamamoto, A. Analysis of a recycling-impaired mutant of low density lipoprotein receptor in familial hypercholesterolemia. J. Biol. Chem. 1989, 264, 16584–16590. [Google Scholar] [CrossRef]
- Agirbasli, D.; Hyatt, T.; Agirbasli, M. Familial hypercholesterolemia with extensive coronary artery disease and tuberous and tendinous xanthomas: A case report and mutation analysis. J. Clin. Lipidol. 2018, 12, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.B.; Horst, M.; Young, M.; Pascua, C.; Puffenberger, E.G.; Brigatti, K.W.; Gonzaga-Jauregui, C.; Shuldiner, A.R.; Gidding, S.; Strauss, K.A.; et al. Clinical characterization of familial hypercholesterolemia due to an amish founder mutation in Apolipoprotein B. BMC Cardiovasc. Disord. 2022, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Petrulioniene, Z.; Gargalskaite, U.; Mikstiene, V.; Norvilas, R.; Skiauteryte, E.; Utkus, A. Autosomal recessive hypercholesterolemia: Case report. J. Clin. Lipidol. 2019, 13, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.F.; Bordoni, B. Hyperlipidemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559182/ (accessed on 9 March 2025).
- Noubiap, J.J.; Nyaga, U.F. Cardiovascular disease prevention should start in early life. BMC Glob. Public Health 2023, 1, 14. [Google Scholar] [CrossRef]
- Rogozik, J.; Główczyńska, R.; Grabowski, M. Genetic backgrounds and diagnosis of familial hypercholesterolemia. Clin. Genet. 2024, 105, 3–12. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Harada-Shiba, M.; Ohtake, A.; Sugiyama, D.; Tada, H.; Dobashi, K.; Matsuki, K.; Minamino, T.; Yamashita, S.; Yamamoto, Y. Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022. J. Atheroscler. Thromb. 2023, 30, 531–557. [Google Scholar] [CrossRef]
- Huijgen, R.; Hutten, B.A.; Kindt, I.; Vissers, M.N.; Kastelein, J.J.P. Discriminative Ability of LDL-Cholesterol to Identify Patients With Familial Hypercholesterolemia. Circ. Cardiovasc. Genet. 2012, 5, 354–359. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Hegele, R.A. Role of DNA copy number variation in dyslipidemias. Curr. Opin. Lipidol. 2018, 29, 125–132. [Google Scholar] [CrossRef]
- Futema, M.; Taylor-Beadling, A.; Williams, M.; Humphries, S.E. Genetic testing for familial hypercholesterolemia—Past, present, and future. J. Lipid Res. 2021, 62, 100139. [Google Scholar] [CrossRef]
- Tada, H.; Kojima, N.; Yamagami, K.; Nomura, A.; Nohara, A.; Usui, S.; Sakata, K.; Hayashi, K.; Fujino, N.; Takamura, M.; et al. Impact of variants of uncertain significance of LDL receptor on phenotypes of familial hypercholesterolemia. J. Clin. Lipidol. 2022, 16, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Haralambos, K.; Whatley, S.D.; Edwards, R.; Gingell, R.; Townsend, D.; Holmans, P.; Clarke, A.; Datta, B.N.; McDowell, I.F.W. Genetic variants of uncertain significance (VUS) in familial hypercholesterolaemia (FH): Can family based association studies help determine pathogenicity? Atherosclerosis 2014, 236, e304. [Google Scholar] [CrossRef]
- Vuorio, A.; Kuoppala, J.; Kovanen, P.T.; Humphries, S.E.; Tonstad, S.; Wiegman, A.; Drogari, E.; Ramaswami, U. Statins for children with familial hypercholesterolemia. Cochrane Database Syst. Rev. 2019, 11, CD006401. [Google Scholar] [CrossRef]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; López-Miranda, J.; Padró, T.; Muñiz, O.; Díaz-Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) Levels in Familial Hypercholesterolemia: An Important Predictor of Cardiovascular Disease Independent of the Type of LDL Receptor Mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef]
- Alieva, A.; Costanzo, A.D.; Reutova, O.; Usova, E.; Sokolnikova, P.; Bakaleiko, V.; Galimberti, F.; Olmastroni, E.; Kostareva, A.; Catapano, A.; et al. Cascade screening for FH patients with VUS to improve pathogenicity classification. Atherosclerosis 2024, 395, 118217. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, C.; de la Peña, G.; Sanguino, J.; Poyatos-Peláez, S.; Carazo, A.; Martínez-Hernández, P.L.; Arrieta, F.; Mostaza, J.M.; Gómez-Coronado, D.; Rodríguez-Nóvoa, S. Identification and Functional Analysis of APOB Variants in a Cohort of Hypercholesterolemic Patients. Int. J. Mol. Sci. 2023, 24, 7635. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.M.; Alves, A.C.; Miranda, B.; Chora, J.R.; Bourbon, M.; Bourbon, M.; Rato, Q.; Alves, A.C.; Medeiros, A.M.; Gomes, A.C.; et al. Unraveling the genetic background of individuals with a clinical familial hypercholesterolemia phenotype. J. Lipid Res. 2024, 65, 100490. [Google Scholar] [CrossRef]
- Tsai, G.J.; Garrett, L.T.; Makhnoon, S.; Bowen, D.J.; Burke, W.; Shirts, B.H. Patient goals, motivations, and attitudes in a patient-driven variant reclassification study. J. Genet. Couns. 2019, 28, 558–569. [Google Scholar] [CrossRef]
- Klevmoen, M.; Mulder, J.W.C.M.; Roeters van Lennep, J.E.; Holven, K.B. Sex Differences in Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2023, 25, 861–868. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Genomic/Variant Coordinates | Pathogenicity (ACMG *) | Number of Carriers | Incidence of ASCVD ** |
|---|---|---|---|---|---|
| LDLR ^ | loss of exons 7–14 | chr19:11221280-11231311 | P ^^ | 5 # (3) ## | 0 |
| LDLR | c.301G>A p.(Glu101Lys) | NM_000527.2:c.301G>A | P | 1 | 0 |
| LDLR | c.654_656del p.(Gly219del) | NM_000527.2:c.654_656del | P | 5 (1) | 0 |
| LDLR | c.910G>A p.(Asp304Asn) | NM_000527.2:c.910G>A | P | 5 (2) | 0 |
| LDLR | c.1187-10G>A | NM_000527.2:c.1187-10G>A | P | 1 | 0 |
| LDLR | c.[1106del];[1106=] p.[(Val369GlyfsTer44)];[(Val369=)] | NM_000527.5:c.[1106del];[1106=] | P | 3 | 0 |
| LDLR | c.[244T>C];[244=] p.[(Cys82Arg)];[(Cys82=)] | NM_000527.5:c.[244T>C];[244=] | P | 1 | 0 |
| LDLR | c.[1013G>A];[1013=] p.[(Cys338Tyr)];[(Cys338=)] | NM_000527.5:c.[1013G>A];[1013=] | P | 1 | 0 |
| LDLR | c.[1775G>A];[1775=] p.[(Gly592Glu)];[(Gly592=)] | NM_000527.5:c.[1775G>A];[1775=] | P | 3 | 1 |
| LDLR | c.[986G>A];[986=] p.[(Cys329Tyr)];[(Cys329=)] | NM_000527.5:c.[986G>A];[986=] | P | 1 | 0 |
| LDLR | c.651_653del p.(Gly219del) | NM_001195798: c.651_653del | P | 1 | 1 |
| LDLR | c.1187-10G>A | NM_000527.2:c.1187-10G>A | P | 1 | 0 |
| APOB § | c.10580G>A p.(Arg3527Gln) | NM_000384.2:c.10580G>A | P | 7 (2) | 2 |
| LDLRAP1 §§ | c.488A>C (p.(Gln163Pro)) | NM_015627.2:c.488A>C | P | 1 (1) | 0 |
| LDLR | c.941-1_946del | NM_000527.2:c.941-1_946del | LP | 6 (2) | 0 |
| LDLR | c.80dup p.(Cys27Trpfs*25) | NM_000527.2:c.80dup | LP | 1 | 0 |
| LDLR | c.986G>A p.(Cys329Tyr) | NM_000527.2:c.986G>A | LP | 3 | 0 |
| LDLR | c.1222G>A p.(Glu408Lys) | NM_000527.2:c.1222G>A | LP | 1 | 0 |
| LDLR | c.418G>A p.(Glu140Lys) | NM_000527.2:c.418G>A | LP | 1 | 0 |
| LDLR | c.1303del p.(Glu435Argfs*16) | NM_000527.2:c.1303del | LP | 1 | 0 |
| LDLR | c.1013G>A p.(Cys338Tyr) | NM_000527.2:c.1013G>A | LP | 1 | 1 |
| LDLR | c. 1027 G>A p.(Gly343Ser) | NM_000527.2:c. 1027 G>A | LP | 1 (1) | 0 |
| LDLR | c.1183del p.(Val395Trpfs*18) | NM_000527.2:c.1183del | LP | 2 (1) | 0 |
| LDLR | c.1217G>A p.(Arg406Gln) | NM_000527.2:c.1217G>A | VUS † | 1 | 0 |
| LDLR | c.1049G>C p.(Arg350Pro) | NM_000527.2:c.1049G>C | VUS | 1 | 1 |
| LDLR | c.1210A>C p.(Thr404Pro) | NM_000527.5:c.1210A>C | VUS | 1 (1) | 0 |
| LDLR | c.58G>A p.(Gly20Arg) | NM_000527.2:c.58G>A | VUS | 1 | 1 |
| LDLR | c.949G>A p.(Glu317Lys) | NM_000527.5:c.949G>A (p.Glu317Lys) | VUS | 1 (1) | 0 |
| APOB | c.4027C>T p.(Pro1343Ser) | NM_000384.2:c.4027C>T | VUS | 1 | 0 |
| APOB | c.7615G>A p.(Val2539Ile) | NM_000384.2:c.7615G>A | VUS | 1 | 0 |
| APOB | c.2630C>T p.(Pro877Leu) | NM_000384.2:c.2630C>T | VUS | 3 | 1 |
| APOB | c.7724A>T p.(Lys2575Ile) | NM_000384.2:c.7724A>T | VUS | 1 | 1 |
| APOB | c.5066G>A p.(Arg1689His) | NM_000384.2:c.5066G>A | VUS | 3 | 1 |
| APOB | c.13480_13482del p.(Gln4494del) | NM_000384.2:c.13480_13482del | VUS | 1 | 0 |
| APOB | c.2248A>G p.(Met750Val) | NM_000384.2:c.2248A>G | VUS | 1 | 0 |
| APOB | c.2450T>C p.(Ile817Thr) | NM_000384.2:c.2450T>C | VUS | 2 (2) | 0 |
| APOB | c.8747C>A p.(Ala2916Asp) | NM_000384.2:c.8747C>A | VUS | 1 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliosaitiene, U.; Cerkauskiene, R.; Laucevicius, A.; Vilniskyte, M.; Sutkus, V.; Mainelis, A.; Burnyte, B.; Barysiene, J.; Petrulioniene, Z. Genetic Spectrum of Lithuanian Familial Hypercholesterolemia Patients. J. Cardiovasc. Dev. Dis. 2025, 12, 197. https://doi.org/10.3390/jcdd12050197
Aliosaitiene U, Cerkauskiene R, Laucevicius A, Vilniskyte M, Sutkus V, Mainelis A, Burnyte B, Barysiene J, Petrulioniene Z. Genetic Spectrum of Lithuanian Familial Hypercholesterolemia Patients. Journal of Cardiovascular Development and Disease. 2025; 12(5):197. https://doi.org/10.3390/jcdd12050197
Chicago/Turabian StyleAliosaitiene, Urte, Rimante Cerkauskiene, Aleksandras Laucevicius, Migle Vilniskyte, Viktoras Sutkus, Antanas Mainelis, Birute Burnyte, Jurate Barysiene, and Zaneta Petrulioniene. 2025. "Genetic Spectrum of Lithuanian Familial Hypercholesterolemia Patients" Journal of Cardiovascular Development and Disease 12, no. 5: 197. https://doi.org/10.3390/jcdd12050197
APA StyleAliosaitiene, U., Cerkauskiene, R., Laucevicius, A., Vilniskyte, M., Sutkus, V., Mainelis, A., Burnyte, B., Barysiene, J., & Petrulioniene, Z. (2025). Genetic Spectrum of Lithuanian Familial Hypercholesterolemia Patients. Journal of Cardiovascular Development and Disease, 12(5), 197. https://doi.org/10.3390/jcdd12050197