
Developmental Aspects of Cardiac Adaptation to Increased Workload
 ,
,  , and
, and 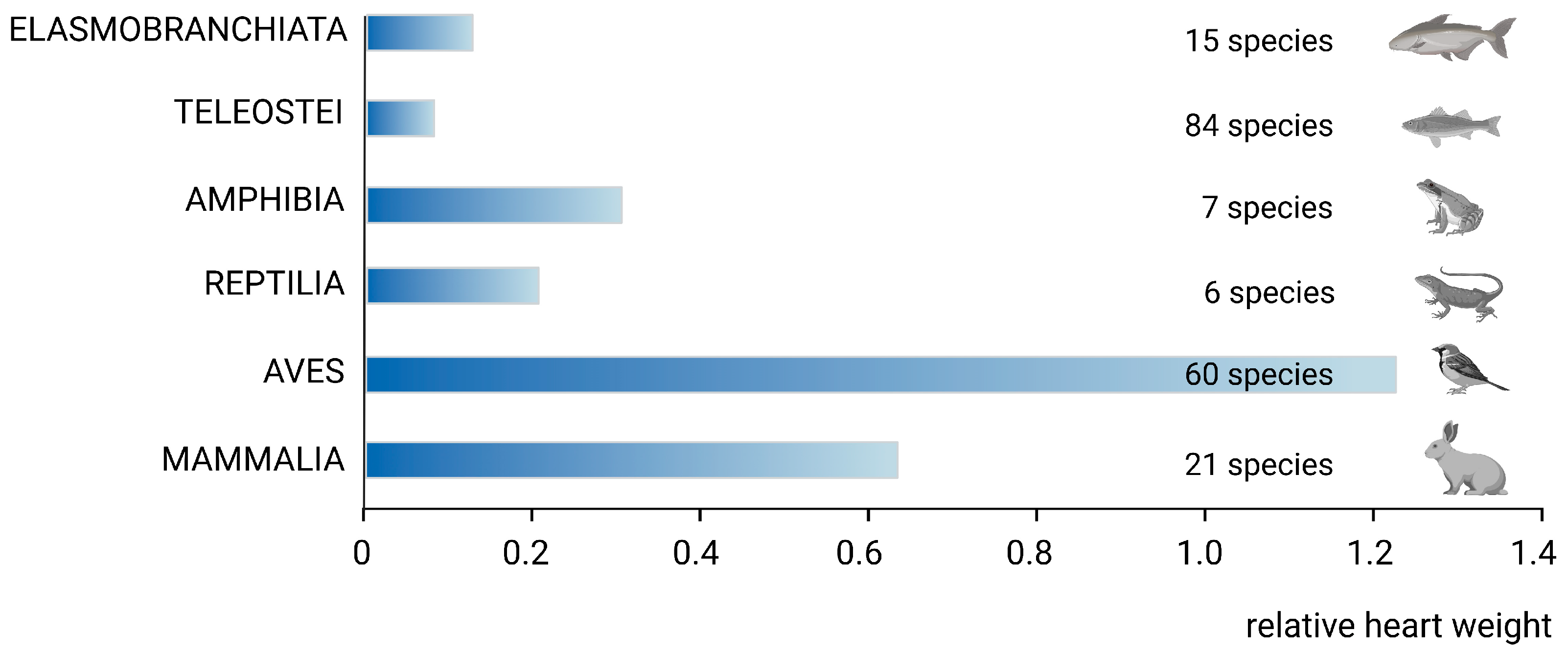{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
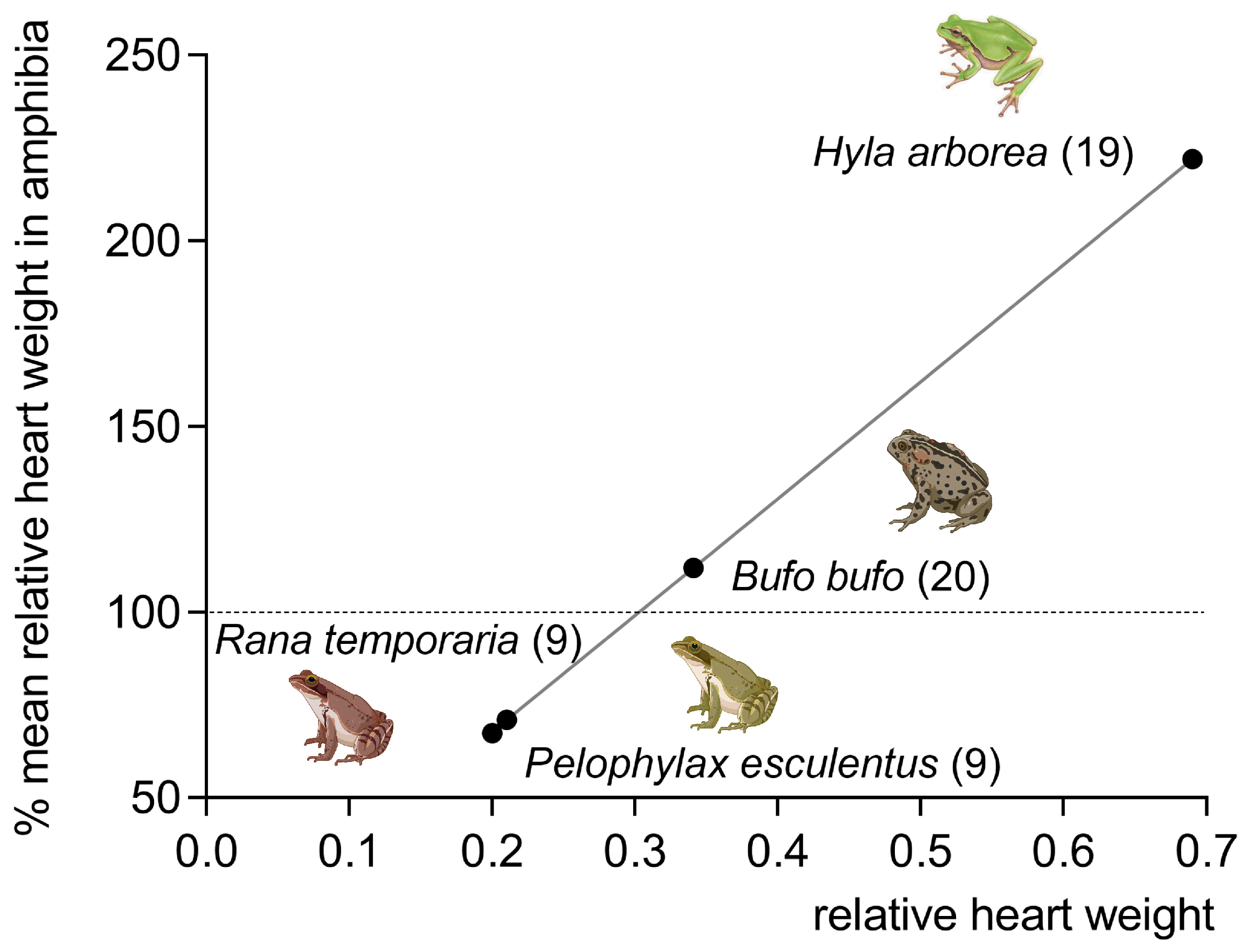
2. Phylogenetic Remarks
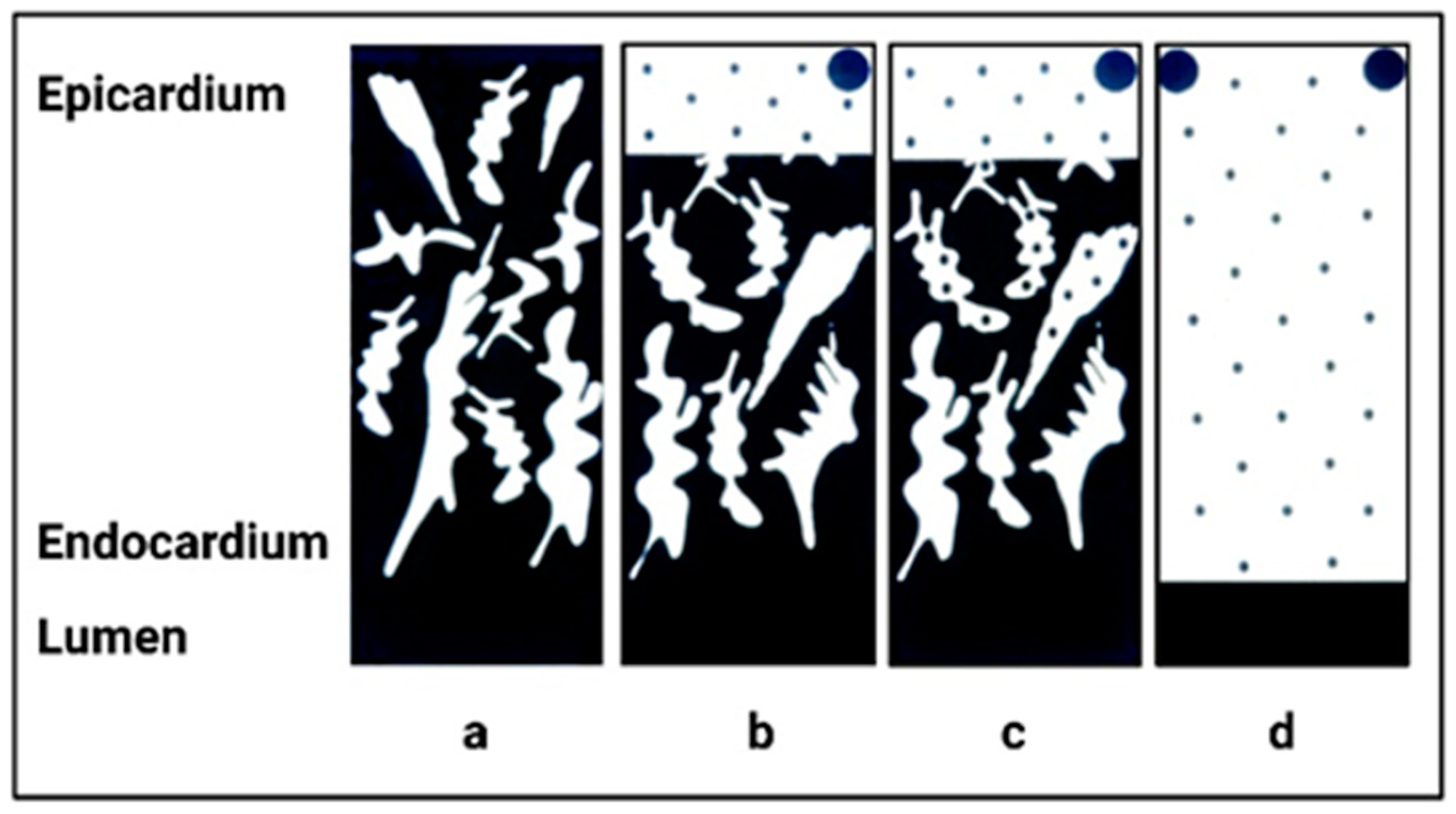
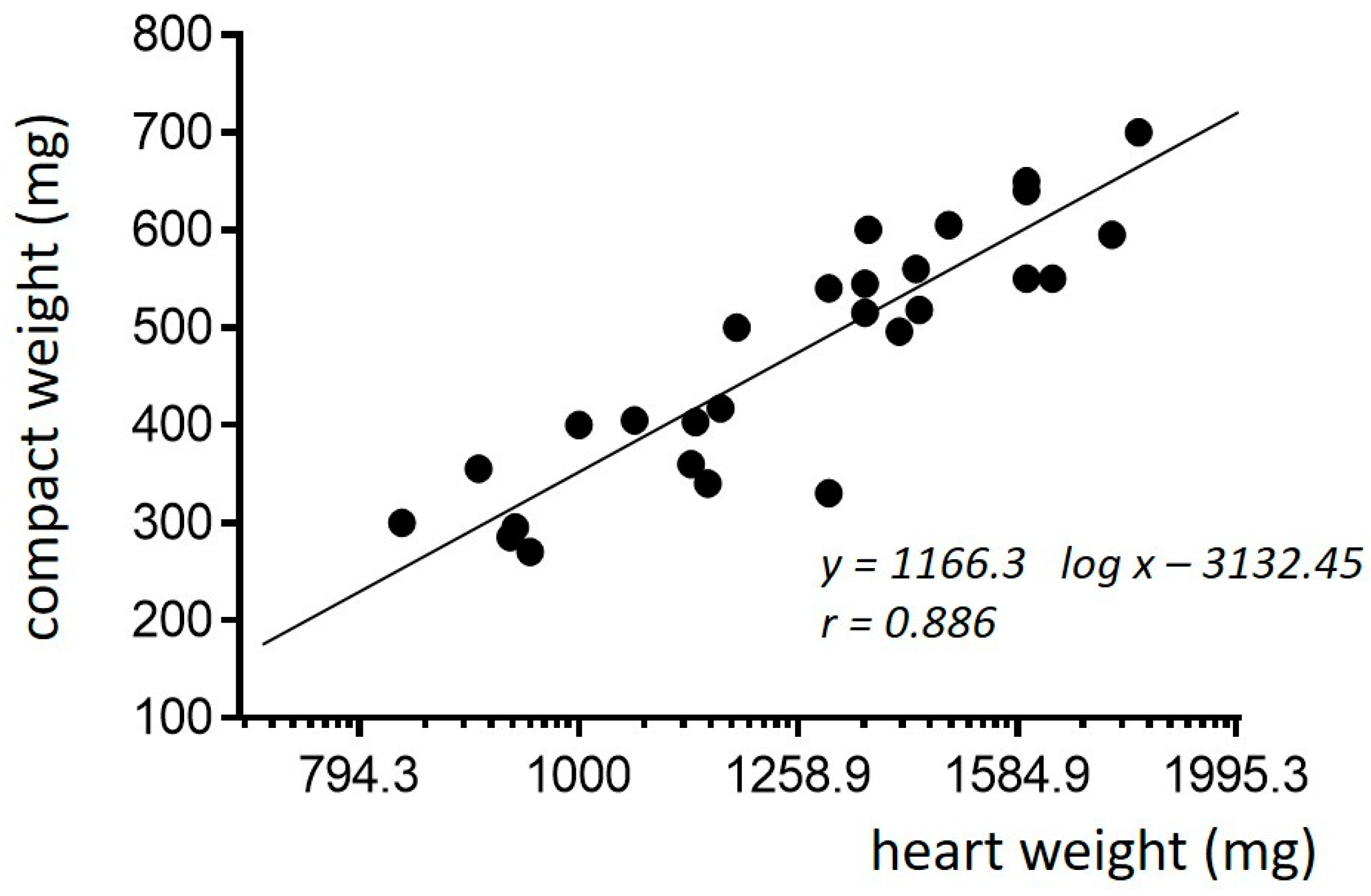
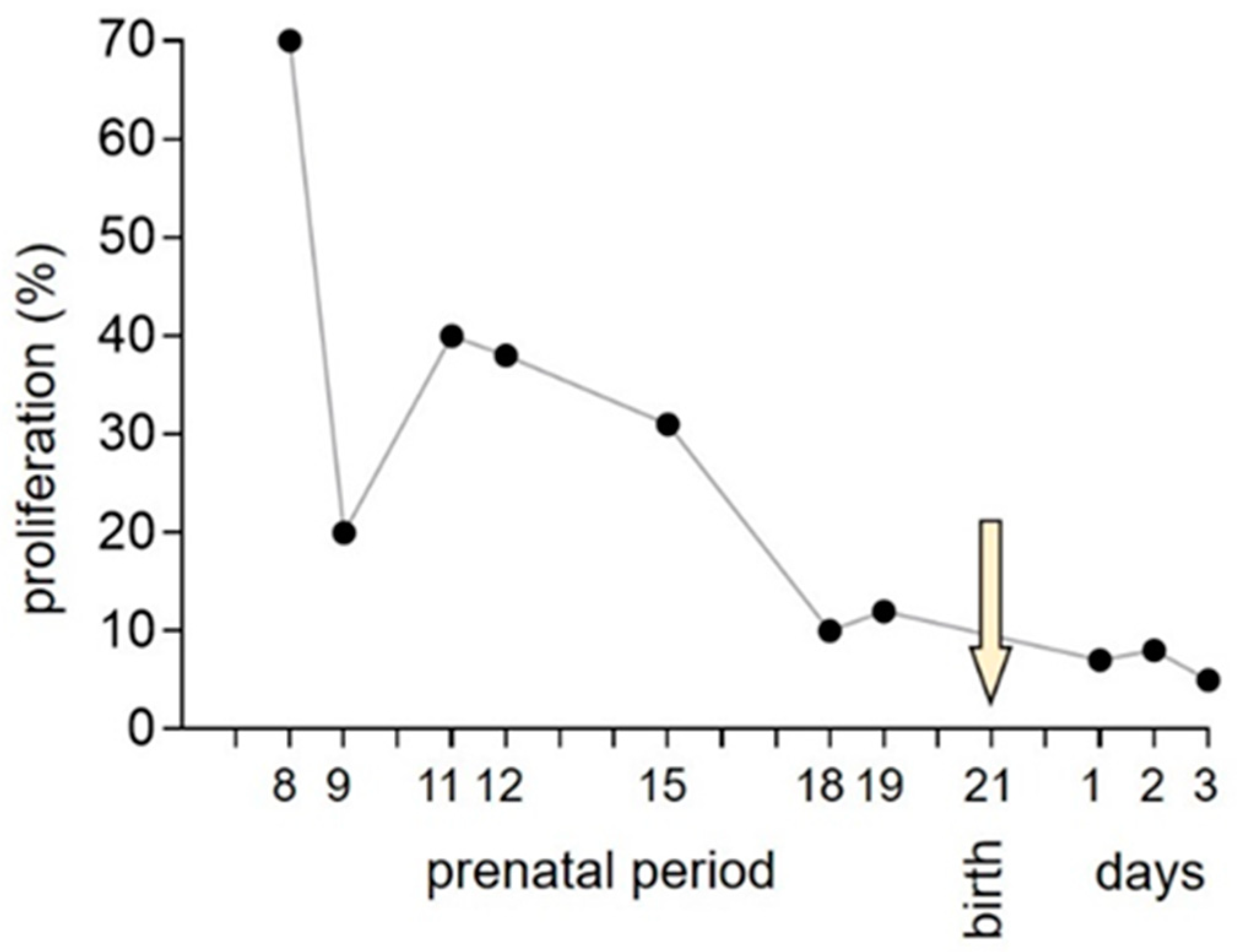
3. Normal Cardiac Growth during Postnatal Ontogeny
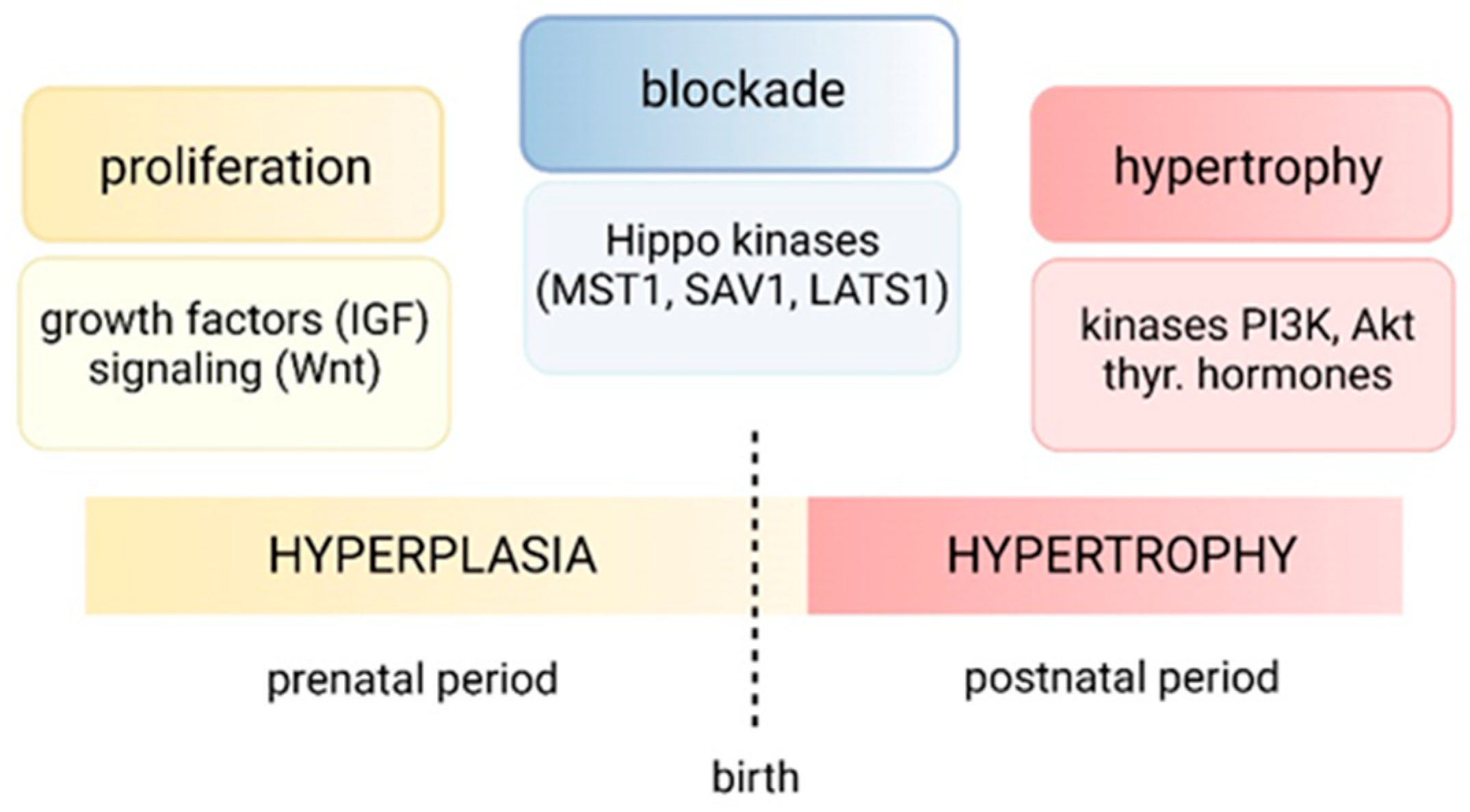
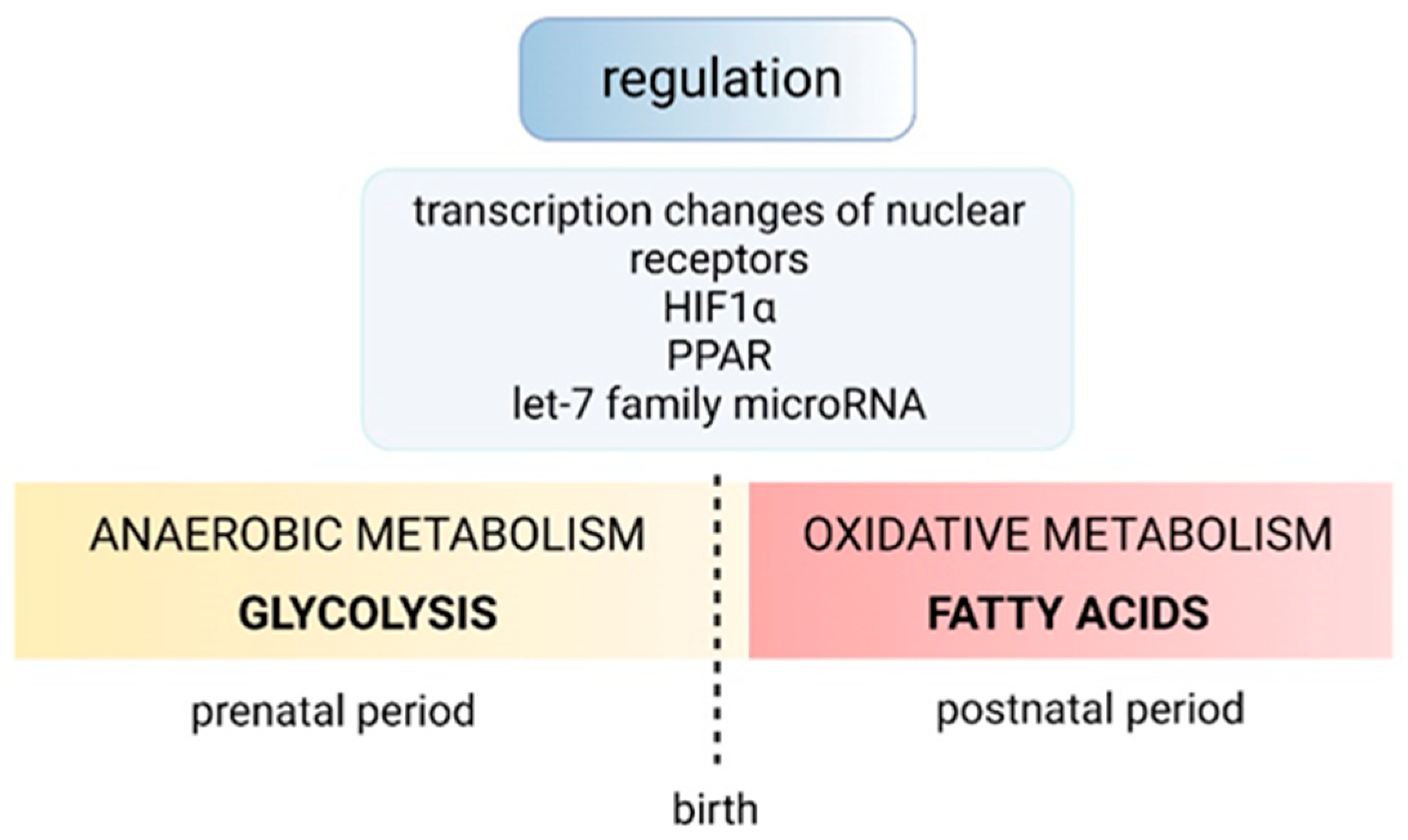
4. Regulation of Normal Cardiac Growth
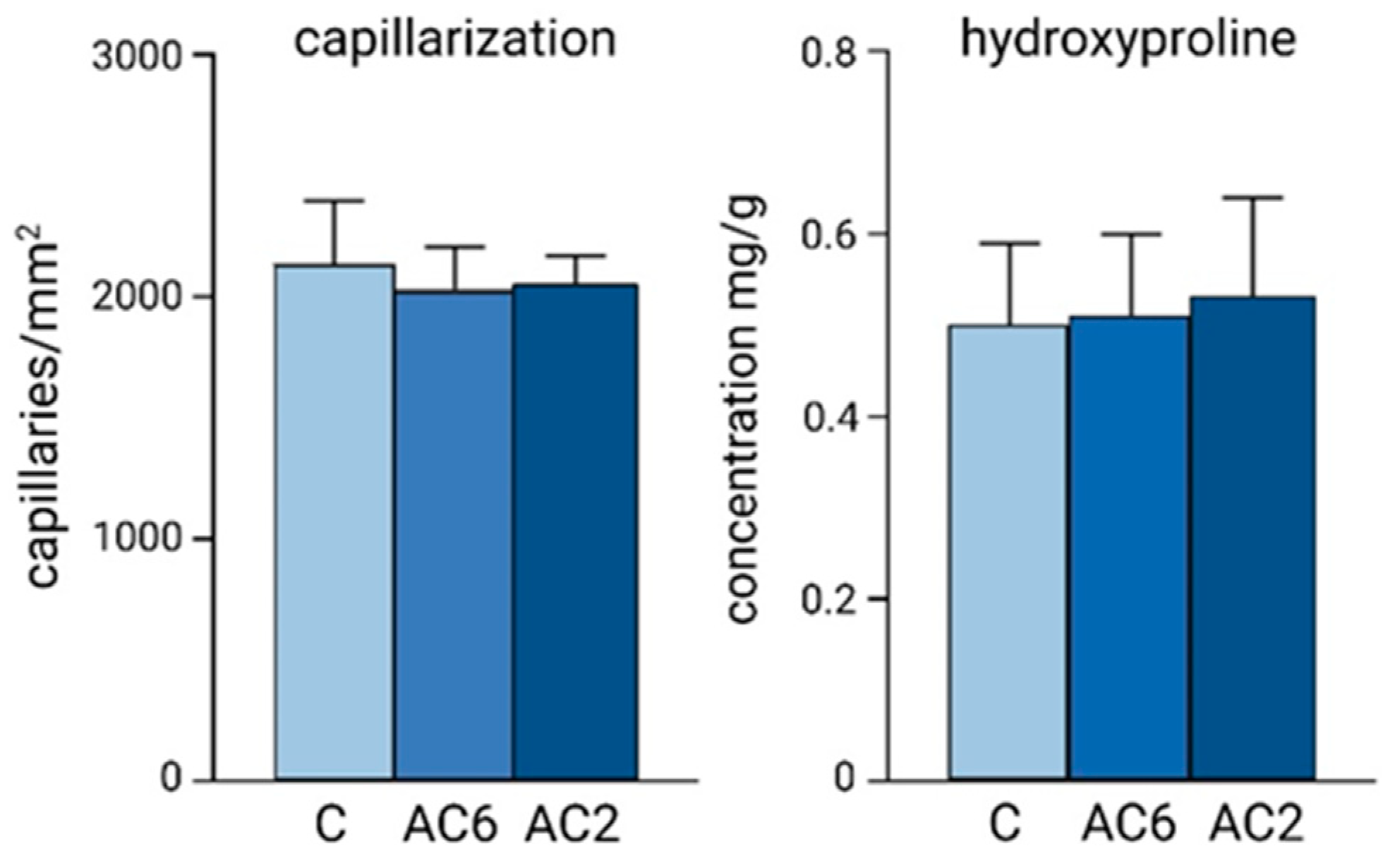
5. Differences in Cardiac Response to the Increased Workload during Postnatal Ontogeny
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Adolph, E.F. General and specific characteristics of physiological adaptations. Am. J. Physiol. 1956, 184, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, B. Comparative aspects of cardiac adaptation. In Cardiac Adaptations; Ostadal, B., Dhalla, N.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–18. [Google Scholar]
- Heallen, T.R.; Kadow, Z.A.; Wang, J.; Martin, J.F. Determinants of Cardiac Growth and Size. Cold Spring Harb. Perspect. Biol. 2020, 12, a037150. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Braun, T. Multimodal Regulation of Cardiac Myocyte Proliferation. Circ. Res. 2017, 121, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; Thompson, R.P. Myocyte proliferation in the developing heart. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2011, 240, 1322–1334. [Google Scholar] [CrossRef]
- Zhu, F.; Meng, Q.; Yu, Y.; Shao, L.; Shen, Z. Adult Cardiomyocyte Proliferation: A New Insight for Myocardial Infarction Therapy. J. Cardiovasc. Transl. Res. 2021, 14, 457–466. [Google Scholar] [CrossRef]
- Hesse, R. Das Herzgewicht der Wirbeltiere. Zool. Jahrb. Abt. Allg. Zool. Physiol. 1921, 38, 243–364. [Google Scholar]
- Poupa, O.; Ostadal, B. Experimental cardiomegalies and “cardiomegalies” in free-living animals. Ann. N. Y. Acad. Sci. 1969, 156, 445–468. [Google Scholar] [CrossRef]
- Clark, A.J. Comparative Physiology of the Heart; Cambridge University Press: Cambridge, UK, 1927. [Google Scholar]
- Poupa, O.; Rakusan, K.; Ostadal, B. The effect of physical activity upon the heart of vertebrates. In Medicine and Sport Science; Karger Publishers: Basel, Switzerland, 1970; Volume 4, pp. 202–233. [Google Scholar]
- Poupa, O. Heart story: A view to the past. In Heart Function in Health and Disease; Ostadal, B., Dhalla, N.S., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1993; pp. 3–22. [Google Scholar]
- Farrell, A.P.; Smith, F. Cardiac form, function and physiology. In The Cardiovascular Systém: Morphology, Control and Function. Fish Physiology; Gamprl, A.K., Gillis, T.E., Farrell, A.P., Brauner, C.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 36A, pp. 155–264. [Google Scholar]
- Jones, D.R.; Brill, R.W.; Bushnell, P.G. Ventricular and Arterial Dynamics of Anaesthetised and Swimming Tuna. J. Exp. Biol. 1993, 182, 97–112. [Google Scholar] [CrossRef]
- Icardo, J.M. Heart morphology and anatomy. In The Cardiovascular Systém: Morphology, Control and Function. Fish Physiology; Gamprl, A.K., Gillis, T.E., Farrell, A.P., Brauner, C.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 36A, pp. 1–47. [Google Scholar]
- Ostadal, B.; Ostadalova, I.; Dhalla, N.S. Development of cardiac sensitivity to oxygen deficiency: Comparative and ontogenetic aspects. Physiol. Rev. 1999, 79, 635–659. [Google Scholar] [CrossRef]
- Ostadal, B.; Schiebler, T.H. The terminal blood bed in the heart of fish. Z. Fur Anat. Entwickl. 1971, 134, 101–110. [Google Scholar]
- Ostadal, B.; Schiebler, T.H.; Rychter, Z. Relations between development of the capillary wall and myoarchitecture of the rat heart. Adv. Exp. Med. Biol. 1975, 53, 375–388. [Google Scholar] [CrossRef]
- Bass, A.; Ostadal, B.; Pelouch, V.; Vitek, V. Differences in weight parameters, myosin-ATPase activity and the enzyme pattern of energy supplying metabolism between the compact and spongious cardiac musculature of carp (Cyprinus carpio) and turtle (Testudo horsfieldi). Pflug. Arch. Eur. J. Physiol. 1973, 343, 65–77. [Google Scholar] [CrossRef]
- Tota, B.; Garofalo, F. Fish heart growth and function: From gross morphology to cell signaling and back. Cardiac nonuniformity: From genes to shape. In Ontogeny and Phylogeny of the Vertebrate Heart; Sedmera, D., Wang, T., Eds.; Springer: New York, NY, USA, 2012; pp. 55–74. [Google Scholar]
- Farrell, A.P.; Farrell, N.D.; Jourdan, H.; Cox, G. A perspective on the evolution of the coronary circulation in fishes and the transition to terrestrial life. In Ontogeny and Phylogeny of the Vertebrate Heart; Sedmera, D., Wang, T., Eds.; Springer: New York, NY, USA, 2012; pp. 75–102. [Google Scholar]
- Ostadal, B.; Rychter, Z.; Poupa, O. Comparative aspects of the development of the terminal vascular bed in the myocardium. Physiol. Bohemoslov. 1970, 19, 1–7. [Google Scholar] [PubMed]
- Becker, R.O.; Chapin, S.; Sherry, R. Regeneration of the ventricular myocardium in amphibians. Nature 1974, 248, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Chablais, F.; Veit, J.; Rainer, G.; Jaźwińska, A. The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 2011, 11, 21. [Google Scholar] [CrossRef]
- Zuppo, D.A.; Tsang, M. Zebrafish heart regeneration: Factors that stimulate cardiomyocyte proliferation. Semin. Cell Dev. Biol. 2020, 100, 3–10. [Google Scholar] [CrossRef]
- Burggren, W.W.; Warburton, S.J. Patterns of form and function in developing hearts: Contributions from non-mammalian vertebrates. Cardioscience 1994, 5, 183–191. [Google Scholar]
- Burggren, W.W. Cardiac design in lower vertebrates: What can phylogeny reveal about ontogeny? Experientia 1988, 44, 919–930. [Google Scholar] [CrossRef]
- Birkedal, R.; Laasmaa, M.; Branovets, J.; Vendelin, M. Ontogeny of cardiomyocytes: Ultrastructure optimization to meet the demand for tight communication in excitation-contraction coupling and energy transfer. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2022, 377, 20210321. [Google Scholar] [CrossRef] [PubMed]
- Rumyantsev, P.P. Cardiomyocytes in the Processes of Reproduction, Differentiation, and Regeneration; Nauka: Leningrad, Russia, 1982. [Google Scholar]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef]
- Rychter, Z.; Rychterova, V.; Lemez, L. Formation of the heart loop and proliferation structure of its wall as a base for ventricular septation. Herz 1979, 4, 86–90. [Google Scholar]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Kajstura, J.; Mansukhani, M.; Cheng, W.; Reiss, K.; Krajewski, S.; Reed, J.C.; Quaini, F.; Sonnenblick, E.H.; Anversa, P. Programmed cell death and expression of the protooncogene bcl-2 in myocytes during postnatal maturation of the heart. Exp. Cell Res. 1995, 219, 110–121. [Google Scholar] [CrossRef]
- Jonker, S.S.; Louey, S.; Giraud, G.D.; Thornburg, K.L.; Faber, J.J. Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4346–4357. [Google Scholar] [CrossRef]
- Anversa, P.; Cheng, W.; Liu, Y.; Leri, A.; Redaelli, G.; Kajstura, J. Apoptosis and myocardial infarction. Basic Res. Cardiol. 1998, 93 (Suppl. S3), 8–12. [Google Scholar] [CrossRef]
- Johnson, J.; Mohsin, S.; Houser, S.R. Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. Int. J. Mol. Sci. 2021, 22, 7764. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Faeh, D.; Moser, A.; Panczak, R.; Bopp, M.; Röösli, M.; Spoerri, A. Independent at heart: Persistent association of altitude with ischaemic heart disease mortality after consideration of climate, topography and built environment. J. Epidemiol. Community Health 2016, 70, 798–806. [Google Scholar] [CrossRef]
- Johnson, J.; Yang, Y.; Bian, Z.; Schena, G.; Li, Y.; Zhang, X.; Eaton, D.M.; Gross, P.; Angheloiu, A.; Shaik, A.; et al. Systemic Hypoxemia Induces Cardiomyocyte Hypertrophy and Right Ventricular Specific Induction of Proliferation. Circ. Res. 2023, 132, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Magadum, A.; Engel, F.B. Cardiomyocyte proliferation in cardiac development and regeneration: A guide to methodologies and interpretations. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1237–H1250. [Google Scholar] [CrossRef]
- Auchampach, J.; Han, L.; Huang, G.N.; Kühn, B.; Lough, J.W.; O’Meara, C.C.; Payumo, A.Y.; Rosenthal, N.A.; Sucov, H.M.; Yutzey, K.E.; et al. Measuring cardiomyocyte cell-cycle activity and proliferation in the age of heart regeneration. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H579–H596. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Engel, F.B. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 67–96. [Google Scholar] [CrossRef]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef]
- Pesevski, Z.; Sedmera, D. Prenatal adaptations to overload. In Cardiac Adaptations; Ostadal, B., Dhalla, N.S., Eds.; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2013; pp. 41–58. [Google Scholar] [CrossRef]
- Krejci, E.; Pesevski, Z.; Nanka, O.; Sedmera, D. Physiological role of FGF signaling in growth and remodeling of developing cardiovascular system. Physiol. Res. 2016, 65, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef]
- Sedmera, D.; Pexieder, T.; Hu, N.; Clark, E.B. Developmental changes in the myocardial architecture of the chick. Anat. Rec. 1997, 248, 421–432. [Google Scholar] [CrossRef]
- Tomanek, R.J. Developmental Progression of the Coronary Vasculature in Human Embryos and Fetuses. Anat. Rec. 2016, 299, 25–41. [Google Scholar] [CrossRef]
- Velkey, J.M.; Bernanke, D.H. Apoptosis during coronary artery orifice development in the chick embryo. Anat. Rec. 2001, 262, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Rakusan, K.; Turek, Z. Protamine inhibits capillary formation in growing rat hearts. Circ. Res. 1985, 57, 393–398. [Google Scholar] [CrossRef]
- Rakusan, K. Cardiac growth, maturation and ageing. In Growth of the Heart in Health and Disease; Zak, R., Ed.; Raven Press: New York, NY, USA, 1984; pp. 131–164. [Google Scholar]
- Lavine, J.S.; Poss, M.; Grenfell, B.T. Directly transmitted viral diseases: Modeling the dynamics of transmission. Trends Microbiol. 2008, 16, 165–172. [Google Scholar] [CrossRef]
- Tomanek, R.J.; Ishii, Y.; Holifield, J.S.; Sjogren, C.L.; Hansen, H.K.; Mikawa, T. VEGF family members regulate myocardial tubulogenesis and coronary artery formation in the embryo. Circ. Res. 2006, 98, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.R.; Kadow, Z.A.; Kim, J.H.; Wang, J.; Martin, J.F. Stimulating Cardiogenesis as a Treatment for Heart Failure. Circ. Res. 2019, 124, 1647–1657. [Google Scholar] [CrossRef]
- Galdos, F.X.; Guo, Y.; Paige, S.L.; VanDusen, N.J.; Wu, S.M.; Pu, W.T. Cardiac Regeneration: Lessons from Development. Circ. Res. 2017, 120, 941–959. [Google Scholar] [CrossRef]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef]
- Leu, M.; Ehler, E.; Perriard, J.C. Characterisation of postnatal growth of the murine heart. Anat. Embryol. 2001, 204, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Collins-Nakai, R.L.; Itoi, T. Developmental changes in energy substrate use by the heart. Cardiovasc. Res. 1992, 26, 1172–1180. [Google Scholar] [CrossRef]
- Dimasi, C.G.; Darby, J.R.T.; Morrison, J.L. A change of heart: Understanding the mechanisms regulating cardiac proliferation and metabolism before and after birth. J. Physiol. 2023, 601, 1319–1341. [Google Scholar] [CrossRef]
- Chung, S.; Dzeja, P.P.; Faustino, R.S.; Perez-Terzic, C.; Behfar, A.; Terzic, A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4 (Suppl. S1), S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Guimarães-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.S.; Chen, J.; McCulloch, A.D.; et al. HIF1α Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef]
- Rakusan, K.; Jelinek, J.; Korecky, B.; Soukupova, M.; Poupa, O. Postnatal development of muscle fibers and capillaries in the rat heart. Physiol. Bohemoslov. 1965, 14, 32–37. [Google Scholar]
- Ostadalova, I.; Kolar, F.; Ostadal, B.; Rohlicek, V.; Rohlicek, J.; Prochazka, J. Early postnatal development of contractile performance and responsiveness to Ca2+, verapamil and ryanodine in the isolated rat heart. J. Mol. Cell. Cardiol. 1993, 25, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Dowell, R.T.; McManus, R.E., 3rd. Pressure-induced cardiac enlargement in neonatal and adult rats. Left ventricular functional characteristics and evidence of cardiac muscle cell proliferation in the neonate. Circ. Res. 1978, 42, 303–310. [Google Scholar] [CrossRef]
- Rakusan, K.; Korecky, B. Regression of cardiomegaly induced in newborn rats. Can. J. Cardiol. 1985, 1, 217–222. [Google Scholar] [PubMed]
- Campbell, S.E.; Rakusan, K.; Gerdes, A.M. Change in cardiac myocyte size distribution in aortic-constricted neonatal rats. Basic Res. Cardiol. 1989, 84, 247–258. [Google Scholar] [CrossRef]
- Campbell, S.E.; Korecky, B.; Rakusan, K. Remodeling of myocyte dimensions in hypertrophic and atrophic rat hearts. Circ. Res. 1991, 68, 984–996. [Google Scholar] [CrossRef]
- Yamamoto, H.; Avkiran, M. Left ventricular pressure overload during postnatal development. Effects on coronary vasodilator reserve and tolerance to hypothermic global ischemia. J. Thorac. Cardiovasc. Surg. 1993, 105, 120–131. [Google Scholar] [CrossRef]
- Kolar, F.; Papousek, F.; Pelouch, V.; Ostadal, B.; Rakusan, K. Pressure overload induced in newborn rats: Effects on left ventricular growth, morphology, and function. Pediatr. Res. 1998, 43, 521–526. [Google Scholar] [CrossRef]
- Sedmera, D.; Thompson, R.P.; Kolar, F. Effect of increased pressure loading on heart growth in neonatal rats. J. Mol. Cell. Cardiol. 2003, 35, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Rakusan, K. Microcirculation in the stressed heart. In The Stressed Heart; Legato, M.J., Ed.; Martinus Nijhoff: Boston, MA, USA, 1987; pp. 107–123. [Google Scholar]
- Weber, K.T.; Brilla, C.G.; Janicki, J.S. Myocardial fibrosis: Functional significance and regulatory factors. Cardiovasc. Res. 1993, 27, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Czubryt, M.P.; Hale, T.M. Cardiac fibrosis: Pathobiology and therapeutic targets. Cell. Signal. 2021, 85, 110066. [Google Scholar] [CrossRef] [PubMed]
- Malek Mohammadi, M.; Abouissa, A.; Azizah, I.; Xie, Y.; Cordero, J.; Shirvani, A.; Gigina, A.; Engelhardt, M.; Trogisch, F.A.; Geffers, R.; et al. Induction of cardiomyocyte proliferation and angiogenesis protects neonatal mice from pressure overload-associated maladaptation. JCI Insight 2019, 4, e128336. [Google Scholar] [CrossRef]
- Malek Mohammadi, M.; Abouissa, A.; Heineke, J. A surgical mouse model of neonatal pressure overload by transverse aortic constriction. Nat. Protoc. 2021, 16, 775–790. [Google Scholar] [CrossRef]
- Hunter, L.E.; Seale, A.N. EDUCATIONAL SERIES IN CONGENITAL HEART DISEASE: Prenatal diagnosis of congenital heart disease. Echo Res. Pract. 2018, 5, R81–R100. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostadal, B.; Kolar, F.; Ostadalova, I.; Sedmera, D.; Olejnickova, V.; Hlavackova, M.; Alanova, P. Developmental Aspects of Cardiac Adaptation to Increased Workload. J. Cardiovasc. Dev. Dis. 2023, 10, 205. https://doi.org/10.3390/jcdd10050205
Ostadal B, Kolar F, Ostadalova I, Sedmera D, Olejnickova V, Hlavackova M, Alanova P. Developmental Aspects of Cardiac Adaptation to Increased Workload. Journal of Cardiovascular Development and Disease. 2023; 10(5):205. https://doi.org/10.3390/jcdd10050205
Chicago/Turabian StyleOstadal, Bohuslav, Frantisek Kolar, Ivana Ostadalova, David Sedmera, Veronika Olejnickova, Marketa Hlavackova, and Petra Alanova. 2023. "Developmental Aspects of Cardiac Adaptation to Increased Workload" Journal of Cardiovascular Development and Disease 10, no. 5: 205. https://doi.org/10.3390/jcdd10050205
APA StyleOstadal, B., Kolar, F., Ostadalova, I., Sedmera, D., Olejnickova, V., Hlavackova, M., & Alanova, P. (2023). Developmental Aspects of Cardiac Adaptation to Increased Workload. Journal of Cardiovascular Development and Disease, 10(5), 205. https://doi.org/10.3390/jcdd10050205