Studies of Genes Involved in Congenital Heart Disease

 and
and
Abstract
:1. Introduction
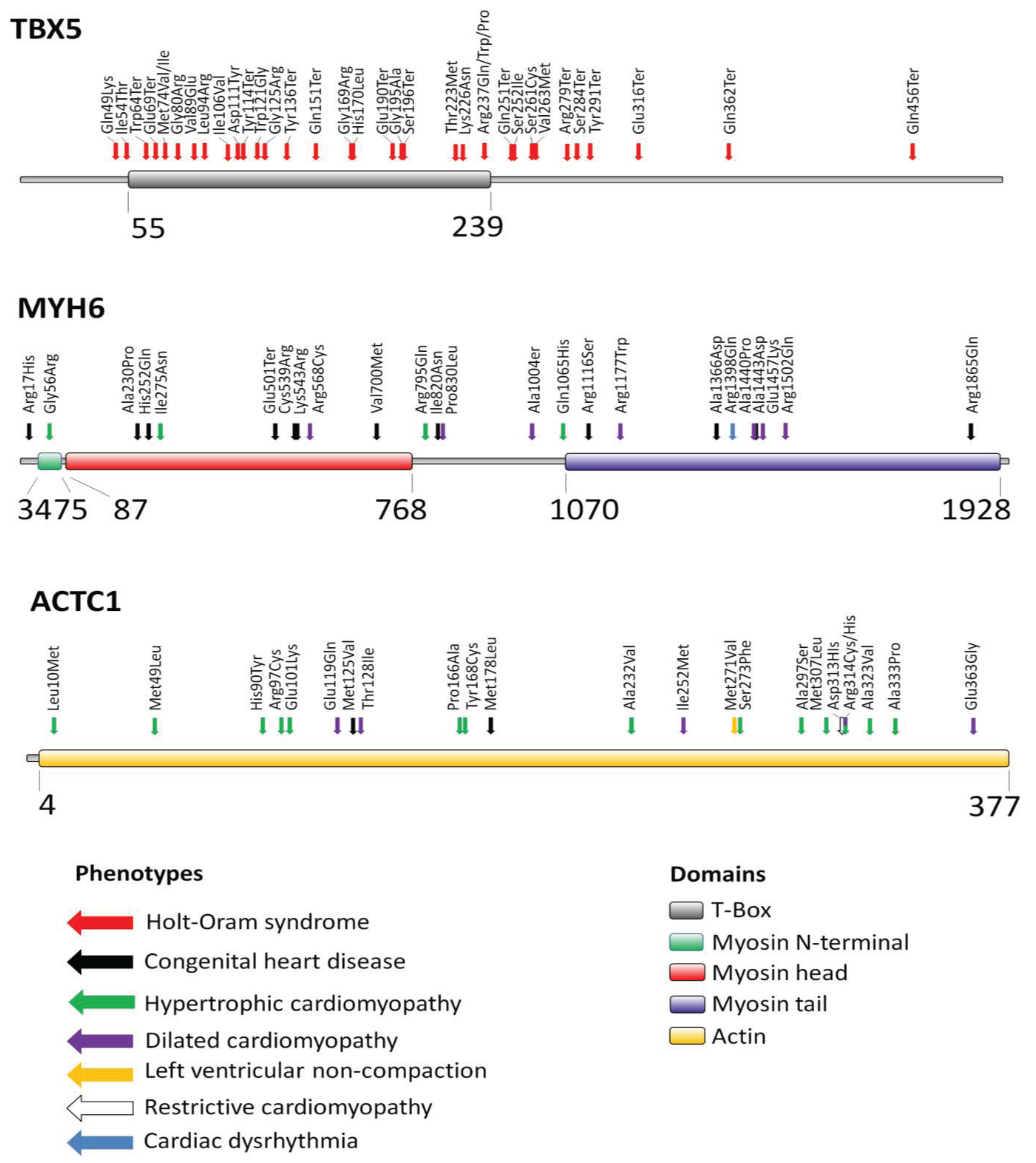
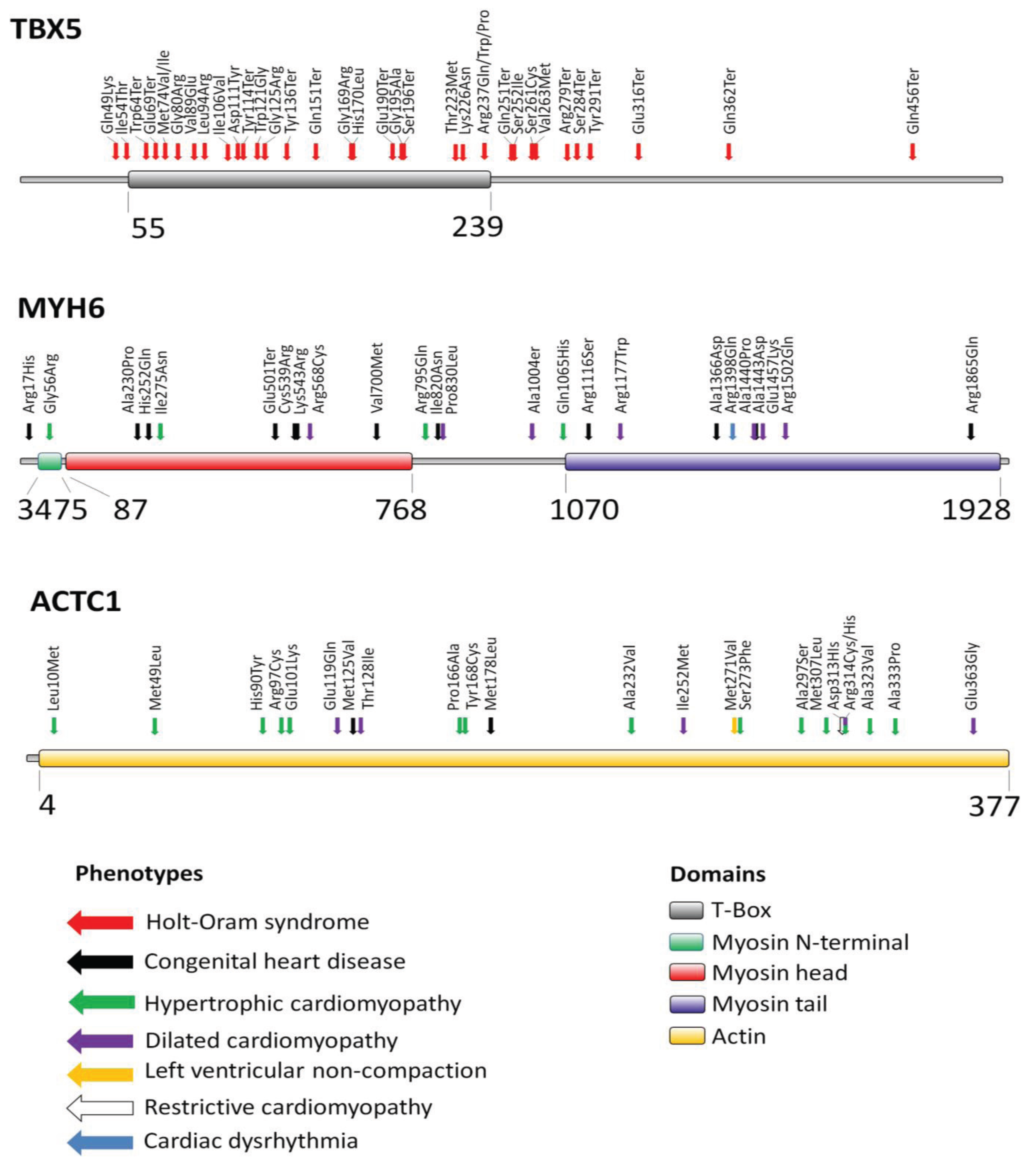
2. Transcription Factor TBX5
3. Myosin Heavy Chain Alpha (MYH6)

{kind=link}
{kind=link}
| Gene | CHD type | Reference |
|---|---|---|
| Transcription factors | ||
| TBX5 | ASD, VSD, CD (Holt-oram syndrome) | [6] |
| TBX1 | ASD, VSD, TOF, PA (DiGeorge syndrome) | [15] |
| TBX20 | ASD, VSD, PFO, MVD | [14] |
| GATA4 | ASD, VSD, AVSD | [11] |
| GATA6 | PTA, TOF, ASD | [16] |
| NKX2.5 | ASD, VSD, SVAS, LVH | [10] |
| NKX2.6 | CAT | [17] |
| MEF2C | OFT | [13] |
| CITED2 | ASD, VSD, PS, TOF | [18] |
| IRX4 | VSD | [19] |
| ZIC3 | Heterotaxy associted with CHDs | [20] |
| SALL4 | Okihiro syndrome/VSD | [21] |
| FOXH1 | TOF, TGA, DORV, CAVC, TA | [22] |
| FOXP1 | AVSD and HLV | [23] |
| ZFPM2/FOG2 | TOF | [24] |
| TFAP2B | Char syndrome, PDA | [25] |
| Sarcomeric protein | ||
| MYH6 | ASD | [32] |
| ACTC1 | ASD | [53] |
| MYH7 | VSD, LVNC | [47] |
| MYH11 | VSD, AA | [48] |
| ELN | SVAS, PS, AS | [49] |
4. Cardiac Actin Alpha (ACTC1)
5. Multiple Mutations and Heterozygosity
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L.; American Heart Association Council on Cardiovascular Disease in the Yong. Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: current knowledge: A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Wren, C.; Irving, C.A.; Griffiths, J.A.; O’Sullivan, J.J.; Chaudhari, M.P.; Haynes, S.R.; Smith, J.H.; Hamilton, J.R.; Hasan, A. Mortality in infants with cardiovascular malformations. Eur. J. Pediatr. 2012, 171, 281–287. [Google Scholar] [CrossRef]
- Clark, K.L.; Yutzey, K.E.; Benson, D.W. Transcription factors and congenital heart defects. Annu. Rev. Physiol. 2006, 68, 97–121. [Google Scholar] [CrossRef]
- McCulley, D.J.; Black, B.L. Transcription factor pathways and congenital heart disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar] [CrossRef]
- Li, Q.Y.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.; Yi, C.H.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997, 15, 21–29. [Google Scholar]
- Ghosh, T.K.; Packham, E.A.; Bonser, A.J.; Robinson, T.E.; Cross, S.J.; Brook, J.D. Characterization of the TBX5 binding site and analysis of mutations that cause Holt-Oram syndrome. Hum. Mol. Genet. 2001, 10, 1983–1994. [Google Scholar] [CrossRef]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E.; et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef]
- Schott, J.J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Song, F.F.; Packham, E.A.; Buxton, S.; Robinson, T.E.; Ronksley, J.; Self, T.; Bonser, A.J.; Brook, J.D. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol. Cell Biol. 2009, 29, 2205–2218. [Google Scholar] [CrossRef]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Ishihara, K.; Oda, M.; Makino, S.; Fukuda, K.; Takahashi, T.; Matsuoka, R.; et al. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circul. J.: Off. J. Jpn. Circul. Soc. 2012, 76, 1703–1711. [Google Scholar] [CrossRef]
- Kirk, E.P.; Sunde, M.; Costa, M.W.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [Google Scholar] [CrossRef]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef]
- Heathcote, K.; Braybrook, C.; Abushaban, L.; Guy, M.; Khetyar, M.E.; Patton, M.A.; Carter, N.D.; Scambler, P.J.; Syrris, P. Common arterial trunk associated with a homeodomain mutation of NKX2.6. Hum. Mol. Genet. 2005, 14, 585–593. [Google Scholar] [CrossRef]
- Sperling, S.; Grimm, C.H.; Dunkel, I.; Mebus, S.; Sperling, H.P.; Ebner, A.; Galli, R.; Lehrach, H.; Fusch, C.; Berger, F.; et al. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum. Mutat. 2005, 26, 575–582. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, J.; Su, D.; Pan, H.; Huang, G.; Li, X.; Li, Z.; Shen, A.; Xie, X.; Wang, B.; Ma, X. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 2011, 130, 657–662. [Google Scholar] [CrossRef]
- Ware, S.M.; Peng, J.; Zhu, L.; Fernbach, S.; Colicos, S.; Casey, B.; Towbin, J.; Belmont, J.W. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am. J. Hum. Genet. 2004, 74, 93–105. [Google Scholar] [CrossRef]
- Kohlhase, J.; Heinrich, M.; Schubert, L.; Liebers, M.; Kispert, A.; Laccone, F.; Turnpenny, P.; Winter, R.M.; Reardon, W. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 2002, 11, 2979–2987. [Google Scholar] [CrossRef]
- Roessler, E.; Ouspenskaia, M.V.; Karkera, J.D.; Velez, J.I.; Kantipong, A.; Lacbawan, F.; Bowers, P.; Belmont, J.W.; Towbin, J.A.; Goldmuntz, E.; et al. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am. J. Hum. Genet. 2008, 83, 18–29. [Google Scholar] [CrossRef]
- Chang, S.W.; Mislankar, M.; Misra, C.; Huang, N.; Dajusta, D.G.; Harrison, S.M.; McBride, K.L.; Baker, L.A.; Garg, V. Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum. Mutat. 2013, 34, 1226–1230. [Google Scholar] [CrossRef]
- Pizzuti, A.; Sarkozy, A.; Newton, A.L.; Conti, E.; Flex, E.; Digilio, M.C.; Amati, F.; Gianni, D.; Tandoi, C.; Marino, B.; et al. Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of fallot. Hum. Mutat. 2003, 22, 372–377. [Google Scholar] [CrossRef]
- Satoda, M.; Zhao, F.; Diaz, G.A.; Burn, J.; Goodship, J.; Davidson, H.R.; Pierpont, M.E.; Gelb, B.D. Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus. Nat. Genet. 2000, 25, 42–46. [Google Scholar] [CrossRef]
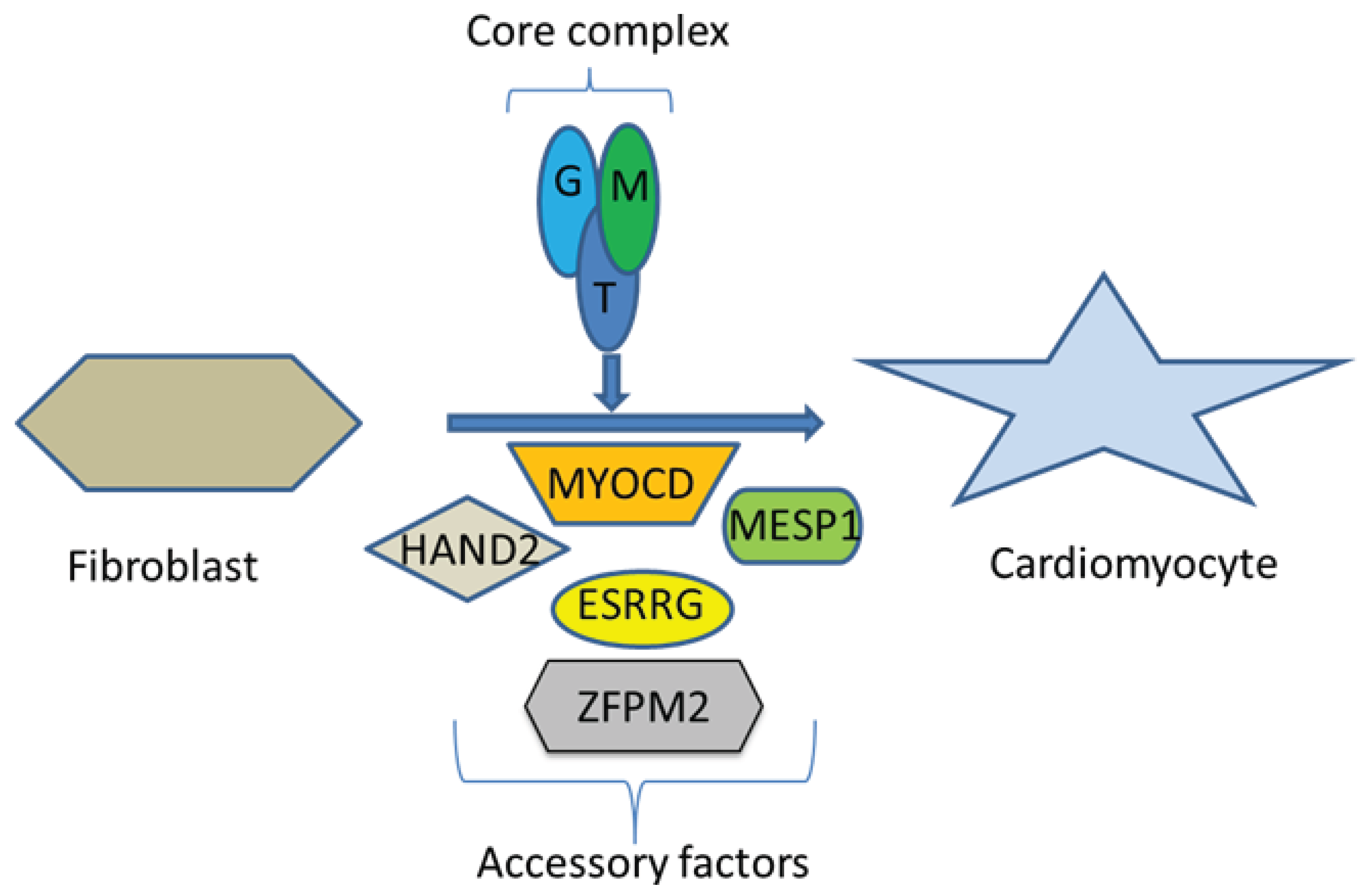
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Fu, J.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef]
- Morimoto, S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc. Res. 2008, 77, 659–666. [Google Scholar] [CrossRef]
- Tanigawa, G.; Jarcho, J.A.; Kass, S.; Solomon, S.D.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. A molecular basis for familial hypertrophic cardiomyopathy: An alpha/beta cardiac myosin heavy chain hybrid gene. Cell 1990, 62, 991–998. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Christe, M.; Conner, D.A.; Ingwall, J.S.; Schoen, F.J.; Seidman, C.E.; Seidman, J.G. A mouse model of familial hypertrophic cardiomyopathy. Science 1996, 272, 731–734. [Google Scholar]
- Ching, Y.H.; Ghosh, T.K.; Cross, S.J.; Packham, E.A.; Honeyman, L.; Loughna, S.; Robinson, T.E.; Dearlove, A.M.; Ribas, G.; Bonser, A.J.; et al. Mutation in myosin heavy chain 6 causes atrial septal defect. Nat. Genet. 2005, 37, 423–428. [Google Scholar] [CrossRef]
- Berdougo, E.; Coleman, H.; Lee, D.H.; Stainier, D.Y.; Yelon, D. Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 2003, 130, 6121–6129. [Google Scholar] [CrossRef]
- Abu-Daya, A.; Sater, A.K.; Wells, D.E.; Mohun, T.J.; Zimmerman, L.B. Absence of heartbeat in the Xenopus tropicalis mutation muzak is caused by a nonsense mutation in cardiac myosin myh6. Dev. Biol. 2009, 336, 20–29. [Google Scholar] [CrossRef]
- Rutland, C.; Warner, L.; Thorpe, A.; Alibhai, A.; Robinson, T.; Shaw, B.; Layfield, R.; Brook, J.D.; Loughna, S. Knockdown of alpha myosin heavy chain disrupts the cytoskeleton and leads to multiple defects during chick cardiogenesis. J. Anat. 2009, 214, 905–915. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Ghosh, T.K.; Pope, M.; Bu’Lock, F.; Thornborough, C.; Eason, J.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; Harvey, R.P.; et al. Alpha-cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Hum. Mol. Genet. 2010, 19, 4007–4016. [Google Scholar] [CrossRef]
- Posch, M.G.; Waldmuller, S.; Muller, M.; Scheffold, T.; Fournier, D.; Andrade-Navarro, M.A.; de Geeter, B.; Guillaumont, S.; Dauphin, C.; Yousseff, D.; et al. Cardiac alpha-myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Arrington, C.B.; Bleyl, S.B.; Matsunami, N.; Bonnell, G.D.; Otterud, B.E.; Nielsen, D.C.; Stevens, J.; Levy, S.; Leppert, M.F.; Bowles, N.E. Exome analysis of a family with pleiotropic congenital heart disease. Circul. Cardiovasc. Genet. 2012, 5, 175–182. [Google Scholar] [CrossRef]
- Niimura, H.; Patton, K.K.; McKenna, W.J.; Soults, J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002, 105, 446–451. [Google Scholar] [CrossRef]
- Carniel, E.; Taylor, M.R.; Sinagra, G.; di Lenarda, A.; Ku, L.; Fain, P.R.; Boucek, M.M.; Cavanaugh, J.; Miocic, S.; Slavov, D.; et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005, 112, 54–59. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circul. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef]
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N.; Hershberger, R.E. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010, 121, 2176–2182. [Google Scholar] [CrossRef]
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Progr. Pediatr. Cardiol. 2011, 31, 39–47. [Google Scholar] [CrossRef]
- Holm, H.; Gudbjartsson, D.F.; Arnar, D.O.; Thorleifsson, G.; Thorgeirsson, G.; Stefansdottir, H.; Gudjonsson, S.A.; Jonasdottir, A.; Mathiesen, E.B.; Njolstad, I.; et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat. Genet. 2010, 42, 117–122. [Google Scholar] [CrossRef]
- Deo, R.; Nalls, M.A.; Avery, C.L.; Smith, J.G.; Evans, D.S.; Keller, M.F.; Butler, A.M.; Buxbaum, S.G.; Li, G.; Miguel Quibrera, P.; et al. Common genetic variation near the connexin-43 gene is associated with resting heart rate in African Americans: A genome-wide association study of 13,372 participants. Heart Rhythm Off. J. Heart Rhythm Soc. 2013, 10, 401–408. [Google Scholar] [CrossRef]
- Holm, H.; Gudbjartsson, D.F.; Sulem, P.; Masson, G.; Helgadottir, H.T.; Zanon, C.; Magnusson, O.T.; Helgason, A.; Saemundsdottir, J.; Gylfason, A.; et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat. Genet. 2011, 43, 316–320. [Google Scholar] [CrossRef]
- Postma, A.V.; van Engelen, K.; van de Meerakker, J.; Rahman, T.; Probst, S.; Baars, M.J.; Bauer, U.; Pickardt, T.; Sperling, S.R.; Berger, F.; et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circul. Cardiovasc. Genet. 2011, 4, 43–50. [Google Scholar] [CrossRef]
- Carey, A.S.; Liang, L.; Edwards, J.; Brandt, T.; Mei, H.; Sharp, A.J.; Hsu, D.T.; Newburger, J.W.; Ohye, R.G.; Chung, W.K.; et al. Effect of copy number variants on outcomes for infants with single ventricle heart defects. Circul. Cardiovasc. Genet. 2013, 6, 444–451. [Google Scholar] [CrossRef]
- Curran, M.E.; Atkinson, D.L.; Ewart, A.K.; Morris, C.A.; Leppert, M.F.; Keating, M.T. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 1993, 73, 159–168. [Google Scholar] [CrossRef]
- Olson, T.M.; Doan, T.P.; Kishimoto, N.Y.; Whitby, F.G.; Ackerman, M.J.; Fananapazir, L. Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2000, 32, 1687–1694. [Google Scholar] [CrossRef]
- Mogensen, J.; Klausen, I.C.; Pedersen, A.K.; Egeblad, H.; Bross, P.; Kruse, T.A.; Gregersen, N.; Hansen, P.S.; Baandrup, U.; Borglum, A.D. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 103, R39–R43. [Google Scholar] [CrossRef]
- Vang, S.; Corydon, T.J.; Borglum, A.D.; Scott, M.D.; Frydman, J.; Mogensen, J.; Gregersen, N.; Bross, P. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005, 272, 2037–2049. [Google Scholar] [CrossRef]
- Matsson, H.; Eason, J.; Bookwalter, C.S.; Klar, J.; Gustavsson, P.; Sunnegardh, J.; Enell, H.; Jonzon, A.; Vikkula, M.; Gutierrez, I.; et al. Alpha-cardiac actin mutations produce atrial septal defects. Hum. Mol. Genet. 2008, 17, 256–265. [Google Scholar]
- Ingles, J.; Doolan, A.; Chiu, C.; Seidman, J.; Seidman, C.; Semsarian, C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J. Med. Genet. 2005, 42. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Pope, M.; Bu’Lock, F.A.; Thornborough, C.; Eason, J.; Setchfield, K.; Ketley, A.; Kirk, E.P.; Fatkin, D.; Feneley, M.P.; et al. Combined mutation screening of NKX2–5, GATA4 and TBX5 in congenital heart disease: Multiple heterozygosity and novel mutations. Congenit. Heart Dis. 2012, 7, 151–159. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Brook, J.D. The impact of mechanical forces in heart morphogenesis. Circul. Cardiovasc. Genet. 2012, 5, 132–142. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Brook, J.D. Formation, contraction, and mechanotransduction of myofribrils in cardiac development: Clues from genetics. Biochem. Res. Int. 2012. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ghosh, T.K.; Granados-Riveron, J.T.; Buxton, S.; Setchfield, K.; Loughna, S.; Brook, J.D. Studies of Genes Involved in Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2014, 1, 134-145. https://doi.org/10.3390/jcdd1010134
Ghosh TK, Granados-Riveron JT, Buxton S, Setchfield K, Loughna S, Brook JD. Studies of Genes Involved in Congenital Heart Disease. Journal of Cardiovascular Development and Disease. 2014; 1(1):134-145. https://doi.org/10.3390/jcdd1010134
Chicago/Turabian StyleGhosh, Tushar K., Javier T. Granados-Riveron, Sarah Buxton, Kerry Setchfield, Siobhan Loughna, and J. David Brook. 2014. "Studies of Genes Involved in Congenital Heart Disease" Journal of Cardiovascular Development and Disease 1, no. 1: 134-145. https://doi.org/10.3390/jcdd1010134
APA StyleGhosh, T. K., Granados-Riveron, J. T., Buxton, S., Setchfield, K., Loughna, S., & Brook, J. D. (2014). Studies of Genes Involved in Congenital Heart Disease. Journal of Cardiovascular Development and Disease, 1(1), 134-145. https://doi.org/10.3390/jcdd1010134