
Therapeutic Innovations: Tyrosine Kinase Inhibitors in Cancer



Abstract
:1. Introduction
2. Digital Age in Cellular Biology
3. Disease as a Kinase Dysfunction
4. Kinases as an Attractive Drug Target
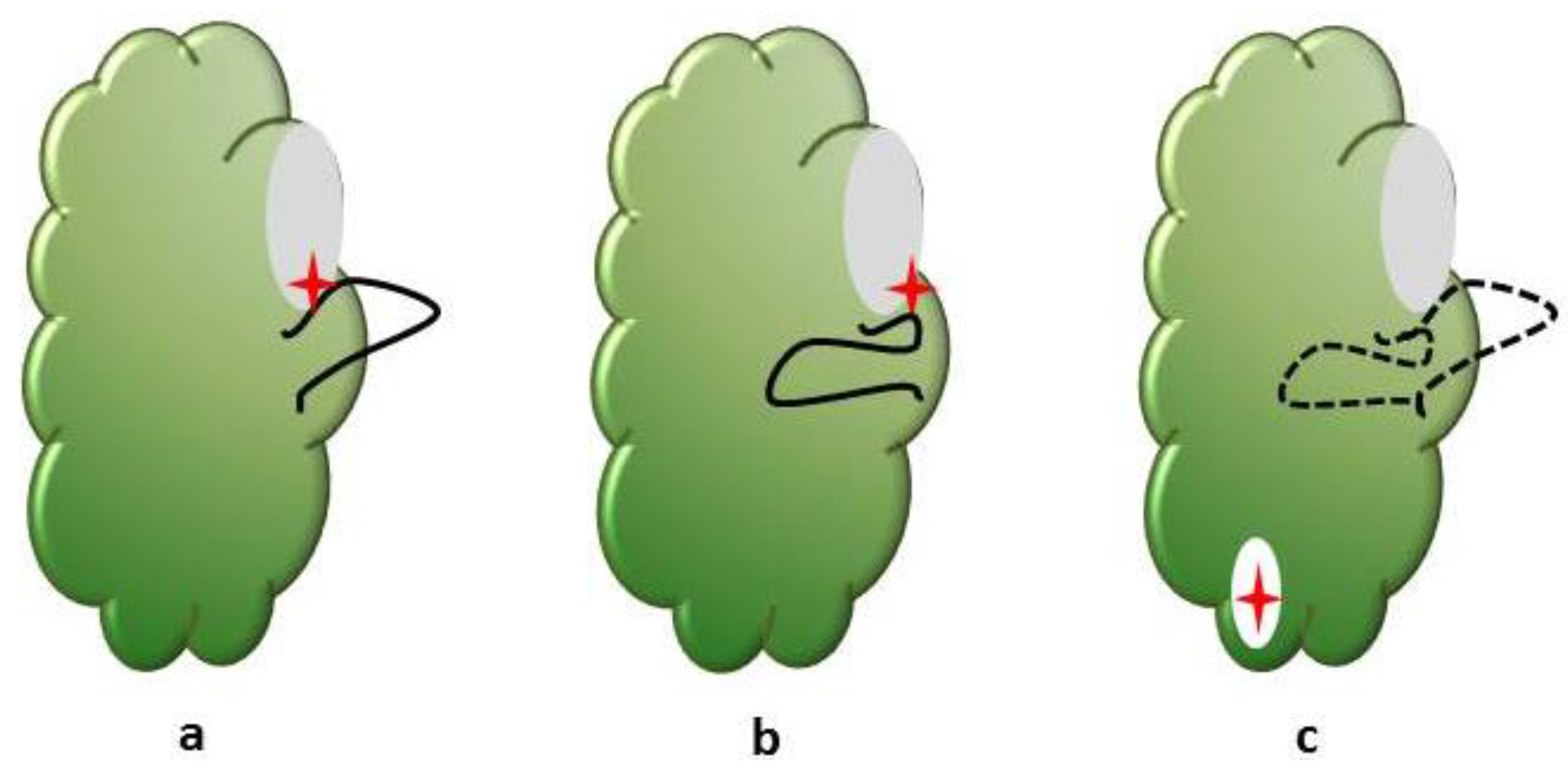
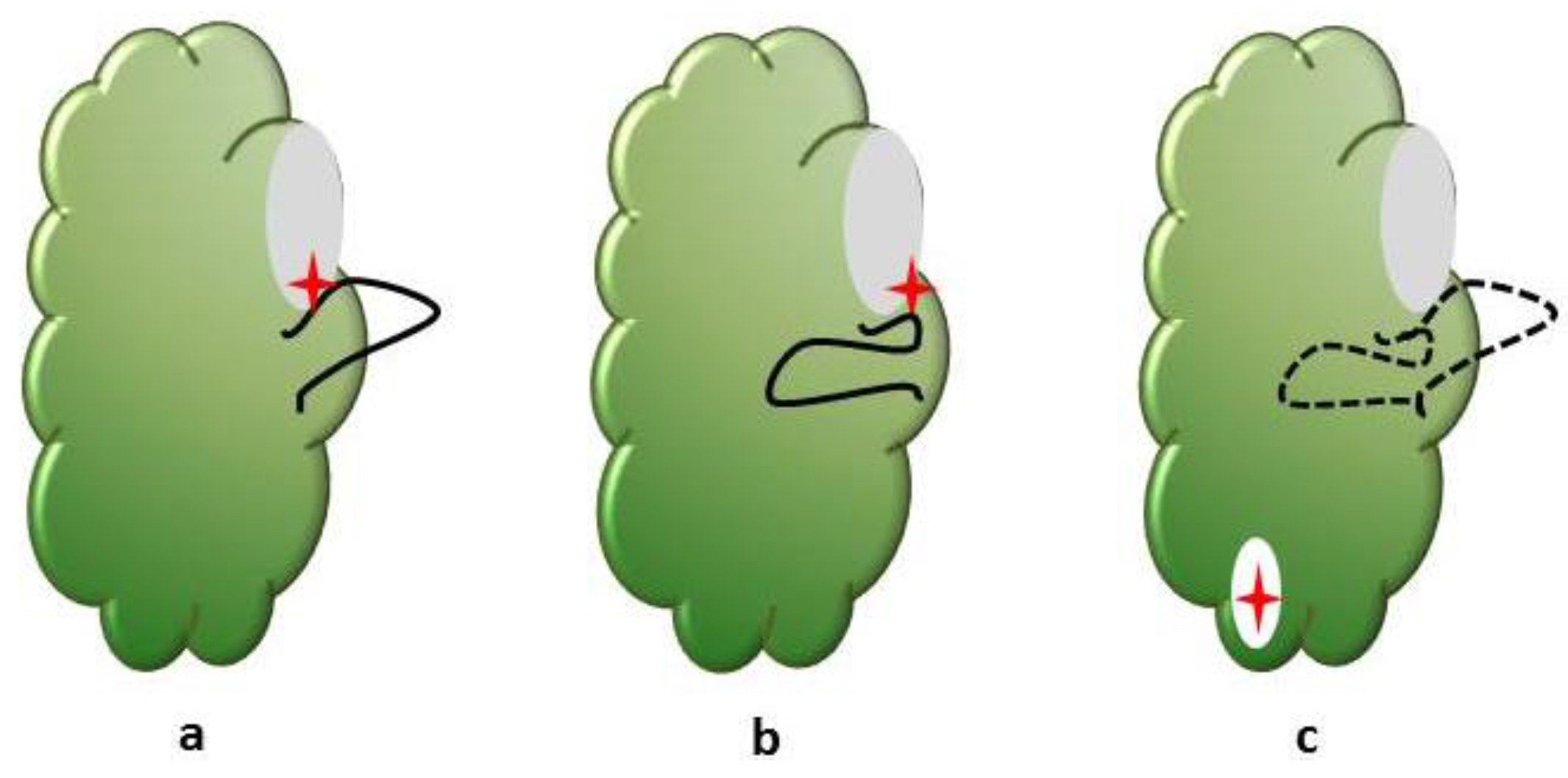
5. Hitting the Target

{kind=link}
| Drug | Target | Year Approved | Indication |
|---|---|---|---|
| Vandetanib (Caprelsa®) | Flt1, Flt4, KDR, EGFR, Ret | 2011 | MTC |
| Crizotinib (Xalkori®) | ALK, MET, EML4-ALK fusion protein | 2011 | NSCLC |
| Ruxolitinib (Jakafi®/Jakavi®) | JAK1, JAK2 | 2011 | Myelofibrosis |
| Vemurafenib (Zelboraf®) | BRAF | 2011 | Melanoma |
| Bosutinib (Bosulif®) | BCR/ABL1 | 2012 | CML |
| Axitinib (Inlyta®) | Flt1, Flt4, KDR, Kit, PDGF-Rα/β | 2012 | RCC |
| Cabozantinib (Cometriq®) | KDR, Mek | 2012 | MTC |
| Regorafinib (Stivarga®) | KDR, TEK | 2012 | CC, GIST |
| Ponatinib (Iclusig®) | BCR/ABL1 | 2012 | CML, ALL |
| Dabrafenib (Tafinlar®) | BRAF | 2013 | Melanoma |
| Trametinib (Mekinist®) | MEK1, MEK2 | 2013 | Melanoma |
| Afatinib (Gilotrif®) | ERBB2, EGFR | 2013 | NSCLC |
| Ibrutinib (Imbruvica®) | BTK | 2013 | MCL, CLL |
| Tofacitinib (Xeljanz®) | JAK3 | 2013 | Rheumatoid arthritis |
| Idelalisib (Zydelig®) | PI3-K | 2014 | CLL |
| Follicular B-cell NHL | |||
| Ceritinib (Zykadia®) | ALK | 2014 | ALK+ NSCLC |
| Lenvatinib (Lenvima®) | VEGFR2 and VEGFR3 | 2015 | Radioactive iodine-refractory DTC |
| Palbociclib (Ibrance®) | CDK4 and CDK6 | 2015 | Breast carcinoma |
| Drug | Targets | Year Approved | Indication |
|---|---|---|---|
| Toceranib (Palladia®) | VEGF-R2 | 2009 | Patnaik grade 2 or 3, recurrent, cutaneous mast cell tumors with or without regional lymph node involvement in dogs |
| PDGF-Rα | |||
| Kit | |||
| Flt-3 | |||
| RET | |||
| JAK family | |||
| Masitinib (Kinavet-CA1®) | Kit | 2010 a | Nonresectable grade 2 and 3 cutaneous mast cell tumors in dogs that have not previously received radiotherapy and/or chemotherapy except corticosteroids |
| PDGF-Rα/β | |||
| Lyn | |||
| FGF-R3 | |||
| Oclacitinib (Apoquel®) | JAK1 | 2013 | Control of pruritus associated with allergic dermatitis and control of atopic dermatitis in dogs at least 12 mos of age |
| JAK 2 |
6. Missing the Point
7. Unanswered Questions
- I
- When to treat with kinase inhibitors?
- II
- What is the biologically sound way to combine kinase inhibitors with cytotoxic chemotherapy?
- III
- How to combine multiple kinase inhibitors in a treatment protocol
- IV
- How to determine efficacy when tumor size is not an appropriate measure
- V
- How to manage treatment failure? Switch to different kinase inhibitors, or add different kinase inhibitors to the current therapy?
8. Future Directions
9. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Tsatsanis, C.; Spandidos, D.A. Oncogenic kinase signaling in human neoplasms. Ann. N. Y. Acad. Sci. 2004, 1028, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A. Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 2001, 101, 2271–2290. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Zhang, J.; Fabbro, D.; Schioth, H.B. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol. Sci. 2014, 35, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar] [PubMed]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, 127–130. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, 204–212. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Muenke, M.; Schell, U.; Hehr, A.; Robin, N.H.; Losken, H.W.; Schinzel, A.; Pulleyn, L.J.; Rutland, P.; Reardon, W.; Malcolm, S.; et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet. 1994, 8, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Acierno, J.S., Jr.; Meysing, A.; Eliseenkova, A.V.; Ma, J.; Ibrahimi, O.A.; Metzger, D.L.; Hayes, F.J.; Dwyer, A.A.; Hughes, V.A.; et al. Mutations in fibroblast growth factor receptor 1 cause both kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2006, 103, 6281–6286. [Google Scholar] [CrossRef] [PubMed]
- CDC. Leading Causes of Death. Available online: http://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm (accessed on 19 January 2016).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Vlahovic, G.; Crawford, J. Activation of tyrosine kinases in cancer. Oncologist 2003, 8, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, I.; Golden, J. Targeting protein kinases. Nat. Rev. Drug Discov. 2004, 3, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of β-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Frantz, B.; Klatt, T.; Pang, M.; Parsons, J.; Rolando, A.; Williams, H.; Tocci, M.J.; O’Keefe, S.J.; O’Neill, E.A. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry 1998, 37, 13846–13853. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [PubMed]
- Dudley, D.T.; Pang, L.; Decker, S.J.; Bridges, A.J.; Saltiel, A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric AKT (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Amiri, K.I.; Burke, J.R.; Schmid, J.A.; Richmond, A. Bms-345541 targets inhibitor of κβ kinase and induces apoptosis in melanoma: Involvement of nuclear factor κβ and mitochondria pathways. Clin. Cancer Res. 2006, 12, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced alk-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- le Coutre, P.; Tassi, E.; Varella-Garcia, M.; Barni, R.; Mologni, L.; Cabrita, G.; Marchesi, E.; Supino, R.; Gambacorti-Passerini, C. Induction of resistance to the abelson inhibitor sti571 in human leukemic cells through gene amplification. Blood 2000, 95, 1758–1766. [Google Scholar] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Sequist, L.V.; Geater, S.L.; Tsai, C.M.; Mok, T.S.; Schuler, M.; Yamamoto, N.; Yu, C.J.; Ou, S.H.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, And LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Lorenzen, S.; Riera Knorrenschild, J.; Haag, G.M.; Pohl, M.; Thuss-Patience, P.; Bassermann, F.; Helbig, U.; Weissinger, F.; Schnoy, E.; Becker, K.; et al. Lapatinib versus lapatinib plus capecitabine as second-line treatment in human epidermal growth factor receptor 2-amplified metastatic gastro-oesophageal cancer: A randomised phase II trial of the arbeitsgemeinschaft internistische onkologie. Eur. J. Cancer 2015, 51, 569–576. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G.; et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Duyster, J.; Bai, R.Y.; Morris, S.W. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene 2001, 20, 5623–5637. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Peters, S.; Bubendorf, L.; Dafni, U.; Kerr, K.M.; Hager, H.; Soltermann, A.; O’Byrne, K.J.; Dooms, C.; Sejda, A.; et al. Prevalence and clinical outcomes for patients with ALK-positive resected stage I to III adenocarcinoma: Results from the european thoracic oncology platform lungscape project. J. Clin. Oncol. 2014, 32, 2780–2787. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J. Targeting receptor tyrosine kinase met in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Hamilton, A.; Elrick, L.J.; Baird, J.W.; Allan, E.K.; Jordanides, N.; Barow, M.; Mountford, J.C.; Holyoake, T.L. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary cml but does not eliminate the quiescent fraction. Blood 2006, 107, 4532–4539. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Bruggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant BCR-ABL. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Lee, F.Y.; Luo, R.; Jiang, Y.; Donker, M.; Akin, C. Dasatinib (BMS-354825) inhibits kitd816v, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood 2006, 108, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Gumireddy, K.; Reddy, M.V.; Cosenza, S.C.; Boominathan, R.; Baker, S.J.; Papathi, N.; Jiang, J.; Holland, J.; Reddy, E.P. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 2005, 7, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Pellicano, F.; Richmond, L.; Allan, E.K.; Hamilton, A.; Lee, F.Y.; Weinmann, R.; Holyoake, T.L. BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood 2008, 111, 2843–2853. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Workman, P. Exploiting the cancer genome: Strategies for the discovery and clinical development of targeted molecular therapeutics. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 549–573. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G. Economics of new oncology drug development. J. Clin. Oncol. 2007, 25, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. The valley of death in anticancer drug development: A reassessment. Trends Pharmacol. Sci. 2012, 33, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y. How to bridge the “valley of death” between a research discovery and clinical application? Ann. Acad. Med. Singap. 2014, 43, 422–424. [Google Scholar] [PubMed]
- Paoloni, M.; Mazcko, C.; Selting, K.; Lana, S.; Barber, L.; Phillips, J.; Skorupski, K.; Vail, D.; Wilson, H.; Biller, B.; et al. Defining the pharmacodynamic profile and therapeutic index of NHS-IL12 immunocytokine in dogs with malignant melanoma. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.; Webb, C.; Mazcko, C.; Cherba, D.; Hendricks, W.; Lana, S.; Ehrhart, E.J.; Charles, B.; Fehling, H.; Kumar, L.; et al. Prospective molecular profiling of canine cancers provides a clinically relevant comparative model for evaluating personalized medicine (PMed) trials. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.; Lana, S.; Thamm, D.; Mazcko, C.; Withrow, S. The creation of the comparative oncology trials consortium pharmacodynamic core: Infrastructure for a virtual laboratory. Vet. J. 2010, 185, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.C.; Mazcko, C.; Fox, E.; Fan, T.; Lana, S.; Kisseberth, W.; Vail, D.M.; Nuckolls, K.; Osborne, T.; Yalkowsy, S.; et al. Rapamycin pharmacokinetic and pharmacodynamic relationships in osteosarcoma: A comparative oncology study in dogs. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.; Paoloni, M.; Mazcko, C.; Khanna, C. The comparative oncology trials consortium: Using spontaneously occurring cancers in dogs to inform the cancer drug development pathway. PLoS Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Paoloni, M.C.; Tandle, A.; Mazcko, C.; Hanna, E.; Kachala, S.; Leblanc, A.; Newman, S.; Vail, D.; Henry, C.; Thamm, D.; et al. Launching a novel preclinical infrastructure: Comparative oncology trials consortium directed therapeutic targeting of TNFα to cancer vasculature. PLoS ONE 2009, 4, e4972. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Bernabe, L.F.; Barnard, S.; Kisseberth, W.C.; Borgatti, A.; Henson, M.; Wilson, H.; Jensen, K.; Ito, D.; Modiano, J.F.; et al. Preclinical evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 in spontaneous canine cancer: Results of a phase I study. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, M.P.; Lundquist, A.; Pirun, M.; Meadows, J.R.; Zamani, N.; Johnson, J.; Sundstrom, G.; Cook, A.; FitzGerald, M.G.; Swofford, R.; et al. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.K.; Mazcko, C.N.; Khanna, C. Defining the value of a comparative approach to cancer drug development. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Simpson, E.R.; Michalowski, A.M.; Hoover, S.B.; Simpson, R.M. Quantifying histological features of cancer biospecimens for biobanking quality assurance using automated morphometric pattern recognition image analysis algorithms. J. Biomol. Tech. 2011, 22, 108–118. [Google Scholar] [PubMed]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Bushell, K.R.; Kim, Y.; Chan, F.C.; Ben-Neriah, S.; Jenks, A.; Alcaide, M.; Fornika, D.; Grande, B.M.; Arthur, S.; Gascoyne, R.D.; et al. Genetic inactivation of TRAF3 in canine and human B-cell lymphoma. Blood 2015, 125, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Elvers, I.; Turner-Maier, J.; Swofford, R.; Koltookian, M.; Johnson, J.; Stewart, C.; Zhang, C.Z.; Schumacher, S.E.; Beroukhim, R.; Rosenberg, M.; et al. Exome sequencing of lymphomas from three dog breeds reveals somatic mutation patterns reflecting genetic background. Genome Res. 2015, 25, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Kennedy, K.; Shapiro, S.G.; Breen, M. BRAF mutations in canine cancers. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Decker, B.; Parker, H.G.; Dhawan, D.; Kwon, E.M.; Karlins, E.; Davis, B.W.; Ramos-Vara, J.A.; Bonney, P.L.; McNiel, E.A.; Knapp, D.W.; et al. Homologous mutation to human BRAF V600E is common in naturally occurring canine bladder cancer—Evidence for a relevant model system and urine-based diagnostic test. Mol. Cancer Res. 2015, 13, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Butler, D. Translational research: Crossing the valley of death. Nature 2008, 453, 840–842. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, J.; Wang, X.; Shen, X. Important steps to improve translation from medical research to health policy. J. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Gadaleta, C.D.; Patruno, R.; Zizzo, N.; Daidone, M.G.; Hansson, M.G.; Paradiso, A.; Ribatti, D. A model of study for human cancer: Spontaneous occurring tumors in dogs. Biological features and translation for new anticancer therapies. Crit. Rev. Oncol. Hematol. 2013, 88, 187–197. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dervisis, N.; Klahn, S. Therapeutic Innovations: Tyrosine Kinase Inhibitors in Cancer. Vet. Sci. 2016, 3, 4. https://doi.org/10.3390/vetsci3010004
Dervisis N, Klahn S. Therapeutic Innovations: Tyrosine Kinase Inhibitors in Cancer. Veterinary Sciences. 2016; 3(1):4. https://doi.org/10.3390/vetsci3010004
Chicago/Turabian StyleDervisis, Nikolaos, and Shawna Klahn. 2016. "Therapeutic Innovations: Tyrosine Kinase Inhibitors in Cancer" Veterinary Sciences 3, no. 1: 4. https://doi.org/10.3390/vetsci3010004
APA StyleDervisis, N., & Klahn, S. (2016). Therapeutic Innovations: Tyrosine Kinase Inhibitors in Cancer. Veterinary Sciences, 3(1), 4. https://doi.org/10.3390/vetsci3010004