

Preliminary Proteomic and Metabolomic Analyses Reveal Potential Serum Biomarkers for Identifying Alveolar Echinococcosis in Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Establishment of AE Mouse Model
2.2. Sample Collection and Processing
2.3. LC-MS/MS Analysis
2.4. Mass Spectrometry Data Analysis and Protein Quantification
2.5. Procedure for Metabolomic Sample Extraction and Data Preprocessing
2.6. Statistical Analysis
2.7. Data Availability
3. Results
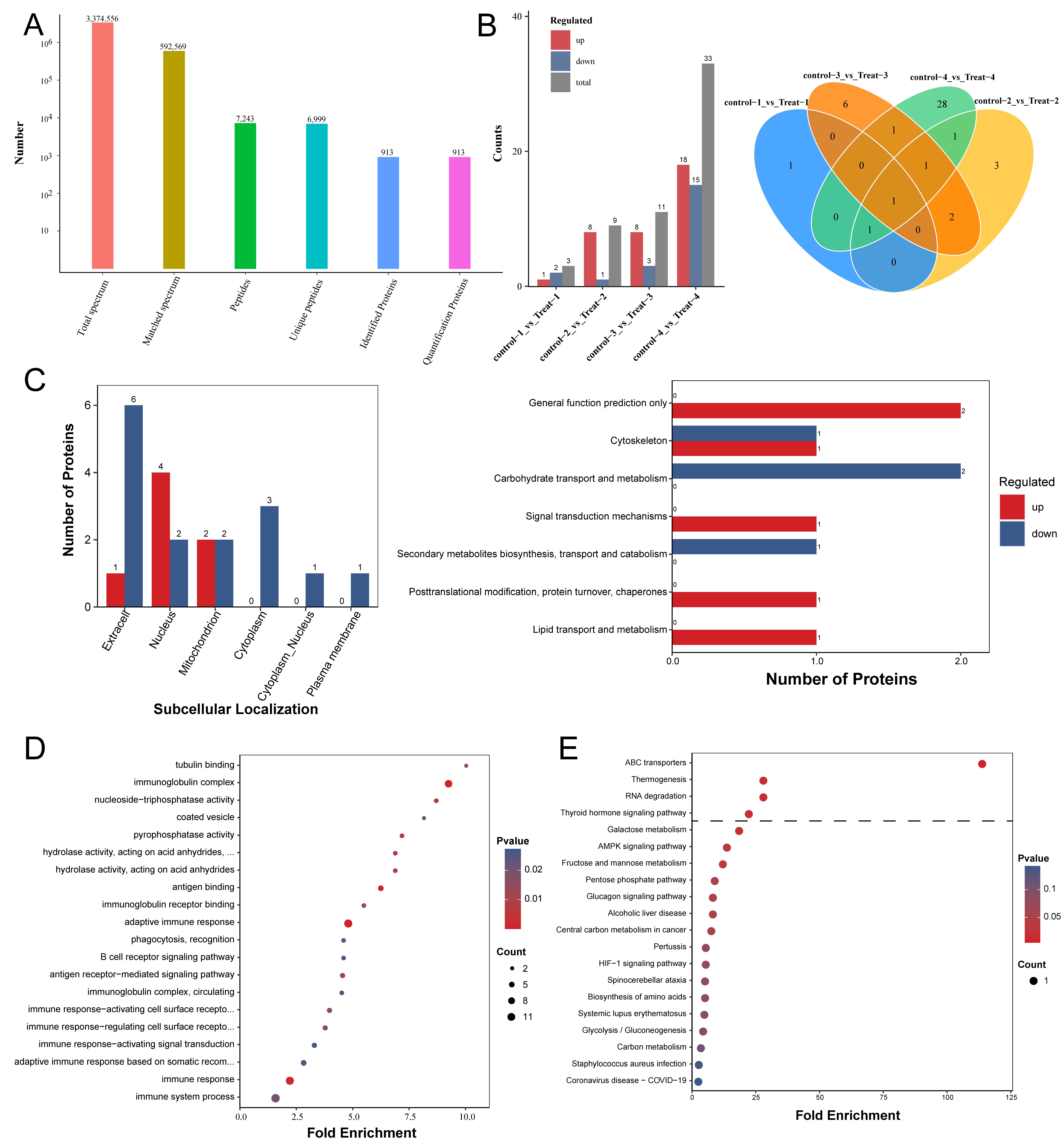
3.1. Differential Serum Protein Profiling and Functional Enrichment Reveal Potential Biomarkers and Key Biological Pathways in Treatment Group
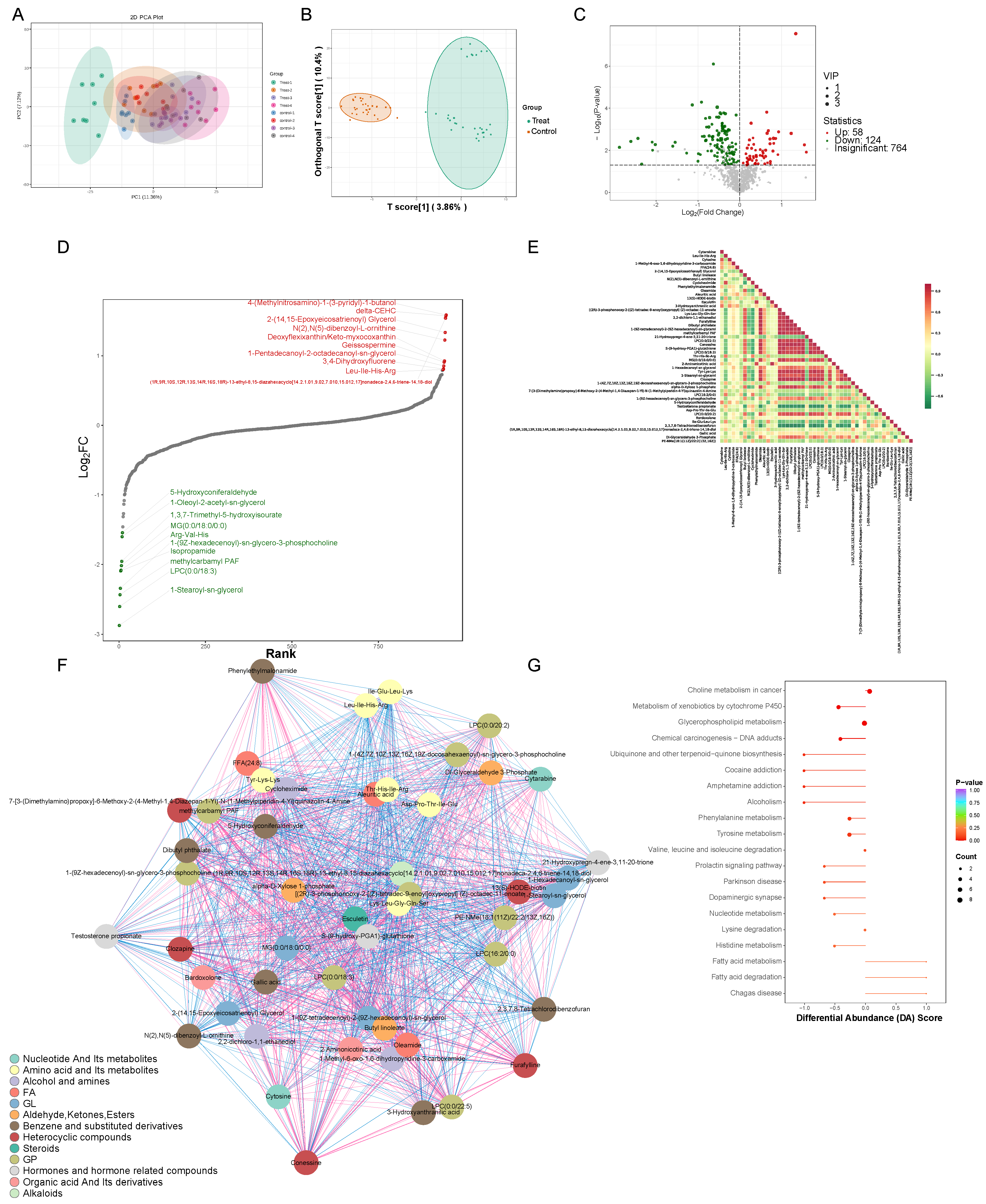
3.2. Untargeted Metabolomics Detected Changes in Mice Serum Metabolites
3.3. Cross-Analysis of Proteomics and Metabolomics Highlights Altered Amino Acid Metabolism in AE-Infected Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wen, H.; Vuitton, L.; Tuxun, T.; Li, J.; Vuitton, D.A.; Zhang, W.; McManus, D.P. Echinococcosis: Advances in the 21st Century. Clin. Microbiol. Rev. 2019, 32. Available online: https://journals.asm.org/doi/full/10.1128/cmr.00075-18 (accessed on 1 May 2025). [CrossRef] [PubMed]
- Romig, T. Epidemiology of echinococcosis. Langenbecks Arch. Surg. 2003, 388, 209–217. [Google Scholar] [CrossRef]
- Nunnari, G.; Pinzone, M.R.; Gruttadauria, S.; Celesia, B.M.; Madeddu, G.; Malaguarnera, G.; Pavone, P.; Cappellani, A.; Cacopardo, B. Hepatic echinococcosis: Clinical and therapeutic aspects. World J. Gastroenterol. 2012, 18, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Piarroux, M.; Piarroux, R.; Giorgi, R.; Knapp, J.; Bardonnet, K.; Sudre, B.; Watelet, J.; Dumortier, J.; Gerard, A.; Beytout, J.; et al. Clinical features and evolution of alveolar echinococcosis in France from 1982 to 2007: Results of a survey in 387 patients. J. Hepatol. 2011, 55, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Romig, T.; Kratzer, W.; Kimmig, P.; Frosch, M.; Gaus, W.; Flegel, W.A.; Gottstein, B.; Lucius, R.; Beckh, K.; Kern, P. An epidemiologic survey of human alveolar echinococcosis in Southwestern Germany. Romerstein Study Group. Am. J. Trop. Med. Hyg. 1999, 61, 566–573. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Wu, W.; Shi, B.; Li, J.; Zhou, X.; Wen, H.; McManus, D.P. Epidemiology and control of echinococcosis in central Asia, with particular reference to the People’s Republic of China. Acta Trop. 2015, 141, 235–243. [Google Scholar] [CrossRef]
- Chong, S.; Chen, G.; Dang, Z.; Niu, F.; Zhang, L.; Ma, H.; Zhao, Y. Echinococcus multilocularis drives the polarization of macrophages by regulating the RhoA-MAPK signaling pathway and thus affects liver fibrosis. Bioengineered 2022, 13, 8747–8758. [Google Scholar] [CrossRef]
- Casulli, A.; Barth, T.F.E.; Tamarozzi, F. Echinococcus multilocularis. Trends Parasitol. 2019, 35, 738–739. [Google Scholar] [CrossRef]
- Thompson, R.C. Biology and Systematics of Echinococcus. Adv. Parasitol. 2017, 95, 65–109. [Google Scholar]
- Muller, J.; Preza, M.; Kaethner, M.; Rufener, R.; Braga, S.; Uldry, A.C.; Heller, M.; Lundstrom-Stadelmann, B. Targeted and non-targeted proteomics to characterize the parasite proteins of Echinococcus multilocularis metacestodes. Front. Cell Infect. Microbiol. 2023, 13, 1170763. [Google Scholar] [CrossRef]
- McManus, D.P.; Gray, D.J.; Zhang, W.; Yang, Y. Diagnosis, treatment, and management of echinococcosis. BMJ 2012, 344, e3866. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, Y.; Zhou, R.; Zhang, H. Suppression of E. multilocularis hydatid cysts after ionizing radiation exposure. PLoS Negl. Trop. Dis. 2013, 7, e2518. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.; Menezes da Silva, A.; Akhan, O.; Mullhaupt, B.; Vizcaychipi, K.A.; Budke, C.; Vuitton, D.A. The Echinococcoses: Diagnosis, Clinical Management and Burden of Disease. Adv. Parasitol. 2017, 96, 259–369. [Google Scholar]
- Kern, P. Clinical features and treatment of alveolar echinococcosis. Curr. Opin. Infect. Dis. 2010, 23, 505–512. [Google Scholar] [CrossRef]
- Bai, Z.; Ma, X.; Yan, R.; Lei, W.; Zhang, Y.; Ren, Y.; Liu, S. Metabolomic profiling of early inactive hepatic alveolar and cystic echinococcosis. Acta Trop. 2023, 242, 106875. [Google Scholar] [CrossRef]
- Ancarola, M.E.; Lichtenstein, G.; Herbig, J.; Holroyd, N.; Mariconti, M.; Brunetti, E.; Berriman, M.; Albrecht, K.; Marcilla, A.; Rosenzvit, M.C.; et al. Extracellular non-coding RNA signatures of the metacestode stage of Echinococcus multilocularis. PLoS Negl. Trop. Dis. 2020, 14, e0008890. [Google Scholar] [CrossRef]
- Cucher, M.A.; Mariconti, M.; Manciulli, T.; Vola, A.; Rosenzvit, M.C.; Brehm, K.; Kamenetzky, L.; Brunetti, E. Circulating Small RNA Profiling of Patients with Alveolar and Cystic Echinococcosis. Biology 2023, 12, 715. [Google Scholar] [CrossRef]
- Ozdemir, S.; Aksungur, N.; Altundas, N.; Kara, S.; Korkut, E.; Ozkaraca, M.; Sefa Mendil, A.; Ozturk, G. Genome-wide profiling of the expression of serum derived exosomal circRNAs in patients with hepatic alveolar echinococcosis. Gene 2022, 814, 146161. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72 e15. [Google Scholar] [CrossRef]
- Qin, G.; Ma, J.; Chen, X.; Chu, Z.; She, Y.M. Methylated-antibody affinity purification to improve proteomic identification of plant RNA polymerase Pol V complex and the interacting proteins. Sci. Rep. 2017, 7, 42943. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.J.; Markwell, J. Assays for determination of protein concentration. Curr. Protoc. Pharmacol. 2007, 38, 3A. [Google Scholar]
- Li, J.; Van Vranken, J.G.; Pontano Vaites, L.; Schweppe, D.K.; Huttlin, E.L.; Etienne, C.; Nandhikonda, P.; Viner, R.; Robitaille, A.M.; Thompson, A.H.; et al. TMTpro reagents: A set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399–404. [Google Scholar] [CrossRef]
- Orsburn, B.C. Proteome Discoverer-A Community Enhanced Data Processing Suite for Protein Informatics. Proteomes 2021, 9, 15. [Google Scholar] [CrossRef]
- Liang, L.; Rasmussen, M.H.; Piening, B.; Shen, X.; Chen, S.; Rost, H.; Snyder, J.K.; Tibshirani, R.; Skotte, L.; Lee, N.C.; et al. Metabolic Dynamics and Prediction of Gestational Age and Time to Delivery in Pregnant Women. Cell 2020, 181, 1680–1692 e1615. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ma, J.; Liu, Y.; Chen, Z.; Xiao, N.; Lu, Y.; Fu, Y.; Yang, C.; Li, M.; Wu, S.; et al. iProX in 2021: Connecting proteomics data sharing with big data. Nucleic Acids Res. 2022, 50, D1522–D1527. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Jin, G.; Guo, N.; Liu, Y.; Zhang, L.; Chen, L.; Dong, T.; Liu, W.; Zhang, X.; Jiang, Y.; Lv, G.; et al. 5-aminolevulinate and CHIL3/CHI3L1 treatment amid ischemia aids liver metabolism and reduces ischemia-reperfusion injury. Theranostics 2023, 13, 4802–4820. [Google Scholar] [CrossRef]
- Torgerson, P.R.; Keller, K.; Magnotta, M.; Ragland, N. The global burden of alveolar echinococcosis. PLoS Negl. Trop. Dis. 2010, 4, e722. [Google Scholar] [CrossRef] [PubMed]
- Lianidou, E.; Pantel, K. Liquid biopsies. Genes. Chromosomes Cancer 2019, 58, 219–232. [Google Scholar] [CrossRef]
- Raza, A.; Khan, A.Q.; Inchakalody, V.P.; Mestiri, S.; Yoosuf, Z.; Bedhiafi, T.; El-Ella, D.M.A.; Taib, N.; Hydrose, S.; Akbar, S.; et al. Dynamic liquid biopsy components as predictive and prognostic biomarkers in colorectal cancer. J. Exp. Clin. Cancer Res. 2022, 41, 99. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Liu, J.; Liu, X.; Tu, Z.; Zheng, Y.; Xu, J.; Fan, H.; Wang, Y.; Hu, M. Multi-omics reveals response mechanism of liver metabolism of hybrid sturgeon under ship noise stress. Sci. Total Environ. 2022, 851, 158348. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.W.; Hojer, C.D.; Zhou, M.; Wu, X.; Wuster, A.; Lee, W.P.; Yaspan, B.L.; Chan, A.C. Regulation of T Cell Receptor Signaling by DENND1B in TH2 Cells and Allergic Disease. Cell 2016, 164, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Ni, W.; Qin, C.; Zhou, Y.; Li, Y.; Huo, J.; Bian, L.; Zhou, A.; Li, J. A functional loop between YTH domain family protein YTHDF3 mediated m(6)A modification and phosphofructokinase PFKL in glycolysis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 334. [Google Scholar] [CrossRef]
- Kalra, S.; Unnikrishnan, A.G.; Baruah, M.P.; Sahay, R.; Bantwal, G. Metabolic and Energy Imbalance in Dysglycemia-Based Chronic Disease. Diabetes Metab. Syndr. Obes. 2021, 14, 165–184. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Yang, Z.; Jiang, Y.; Sun, L.; Huang, D.; Tian, M.; Shen, Y.; Deng, J.; Hou, J.; et al. Echinococcus multilocularis protoscoleces enhance glycolysis to promote M2 Macrophages through PI3K/Akt/mTOR Signaling Pathway. Pathog. Glob. Health 2023, 117, 409–416. [Google Scholar] [CrossRef]
- Crampsie, M.A.; Jones, N.; Das, A.; Aliaga, C.; Desai, D.; Lazarus, P.; Amin, S.; Sharma, A.K. Phenylbutyl isoselenocyanate modulates phase I and II enzymes and inhibits 4-(methylnitrosamino)-1-(3-pyridyl)- 1-butanone-induced DNA adducts in mice. Cancer Prev. Res. 2011, 4, 1884–1894. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Falck, J.R.; Guthi, J.S.; Anjaiah, S.; Capdevila, J.H.; Harris, R.C. Mitogenic activity and signaling mechanism of 2-(14,15-epoxyeicosatrienoyl)glycerol, a novel cytochrome p450 arachidonate metabolite. Mol. Cell Biol. 2007, 27, 3023–3034. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, Y.; Saegusa, N.; Giles, W.R.; Stafforini, D.M.; Spitzer, K.W. Platelet-activating factor stimulates sodium-hydrogen exchange in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2395–H2401. [Google Scholar] [CrossRef]
- Eckert, J.; Deplazes, P. Biological, epidemiological, and clinical aspects of echinococcosis, a zoonosis of increasing concern. Clin. Microbiol. Rev. 2004, 17, 107–135. [Google Scholar] [CrossRef]
- Genchi, G. An overview on D-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- McAllan, L.; Cotter, P.D.; Roche, H.M.; Korpela, R.; Nilaweera, K.N. Impact of leucine on energy balance. J. Physiol. Biochem. 2013, 69, 155–163. [Google Scholar] [CrossRef]
- Zhu, M.; Du, X.; Xu, H.; Yang, S.; Wang, C.; Zhu, Y.; Zhang, T.; Zhao, W. Metabolic profiling of liver and faeces in mice infected with echinococcosis. Parasit. Vectors 2021, 14, 324. [Google Scholar] [CrossRef]
- Ritler, D.; Rufener, R.; Li, J.V.; Kampfer, U.; Muller, J.; Buhr, C.; Schurch, S.; Lundstrom-Stadelmann, B. In vitro metabolomic footprint of the Echinococcus multilocularis metacestode. Sci. Rep. 2019, 9, 19438. [Google Scholar] [CrossRef]
- Cynober, L. Metabolism of Dietary Glutamate in Adults. Ann. Nutr. Metab. 2018, 73 (Suppl. S5), 5–14. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 1–7. [Google Scholar]
- Lin, C.; Chen, Z.; Zhang, L.; Wei, Z.; Cheng, K.K.; Liu, Y.; Shen, G.; Fan, H.; Dong, J. Deciphering the metabolic perturbation in hepatic alveolar echinococcosis: A (1)H NMR-based metabolomics study. Parasit. Vectors 2019, 12, 300. [Google Scholar] [CrossRef]
- Tietge, U.J.; Bahr, M.J.; Manns, M.P.; Boker, K.H. Hepatic amino-acid metabolism in liver cirrhosis and in the long-term course after liver transplantation. Transpl. Int. 2003, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Shamsan, E.; Jiang, B.; Fan, H.; Zhang, Y.; Dehwah, M.A.S. Structural changes and expression of hepatic fibrosis-related proteins in coculture of Echinococcus multilocularis protoscoleces and human hepatic stellate cells. Parasit. Vectors 2021, 14, 593. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, X.; Liu, N.; Liu, J.; Wang, W.; Wang, Y.; Lei, W.; Zhao, C.; Ma, W.; Guo, S.; et al. Preliminary Proteomic and Metabolomic Analyses Reveal Potential Serum Biomarkers for Identifying Alveolar Echinococcosis in Mice. Vet. Sci. 2025, 12, 565. https://doi.org/10.3390/vetsci12060565
Zhang Q, Zhang X, Liu N, Liu J, Wang W, Wang Y, Lei W, Zhao C, Ma W, Guo S, et al. Preliminary Proteomic and Metabolomic Analyses Reveal Potential Serum Biomarkers for Identifying Alveolar Echinococcosis in Mice. Veterinary Sciences. 2025; 12(6):565. https://doi.org/10.3390/vetsci12060565
Chicago/Turabian StyleZhang, Qing, Xiongying Zhang, Na Liu, Jia Liu, Wei Wang, Yongshun Wang, Wen Lei, Cunzhe Zhao, Wanli Ma, Shuai Guo, and et al. 2025. "Preliminary Proteomic and Metabolomic Analyses Reveal Potential Serum Biomarkers for Identifying Alveolar Echinococcosis in Mice" Veterinary Sciences 12, no. 6: 565. https://doi.org/10.3390/vetsci12060565
APA StyleZhang, Q., Zhang, X., Liu, N., Liu, J., Wang, W., Wang, Y., Lei, W., Zhao, C., Ma, W., Guo, S., Cai, H., Zhang, J., Liu, Y., Shi, K., Zhang, W., & Ma, X. (2025). Preliminary Proteomic and Metabolomic Analyses Reveal Potential Serum Biomarkers for Identifying Alveolar Echinococcosis in Mice. Veterinary Sciences, 12(6), 565. https://doi.org/10.3390/vetsci12060565