
Comparative Transcriptomics Analysis of Foot-and-Mouth Disease Virus-Infected Cell Model Systems

Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Virus and Cell Line
2.2. Preparing the Sample
2.3. Analysis of RNA-Seq
2.4. Control of Quality and Mapping of Reads
2.5. Analysis of Differential Expression
2.6. Analyses of GO and KEGG Enrichments
2.7. Analysis of SNP Sites
2.8. Analysis of Differential Exon Usage
2.9. Analysis of Interactions in Protein Networks
2.10. Analysis of GSEA Enrichments
2.11. Validation of Gene Expression by RT-qPCR
3. Results
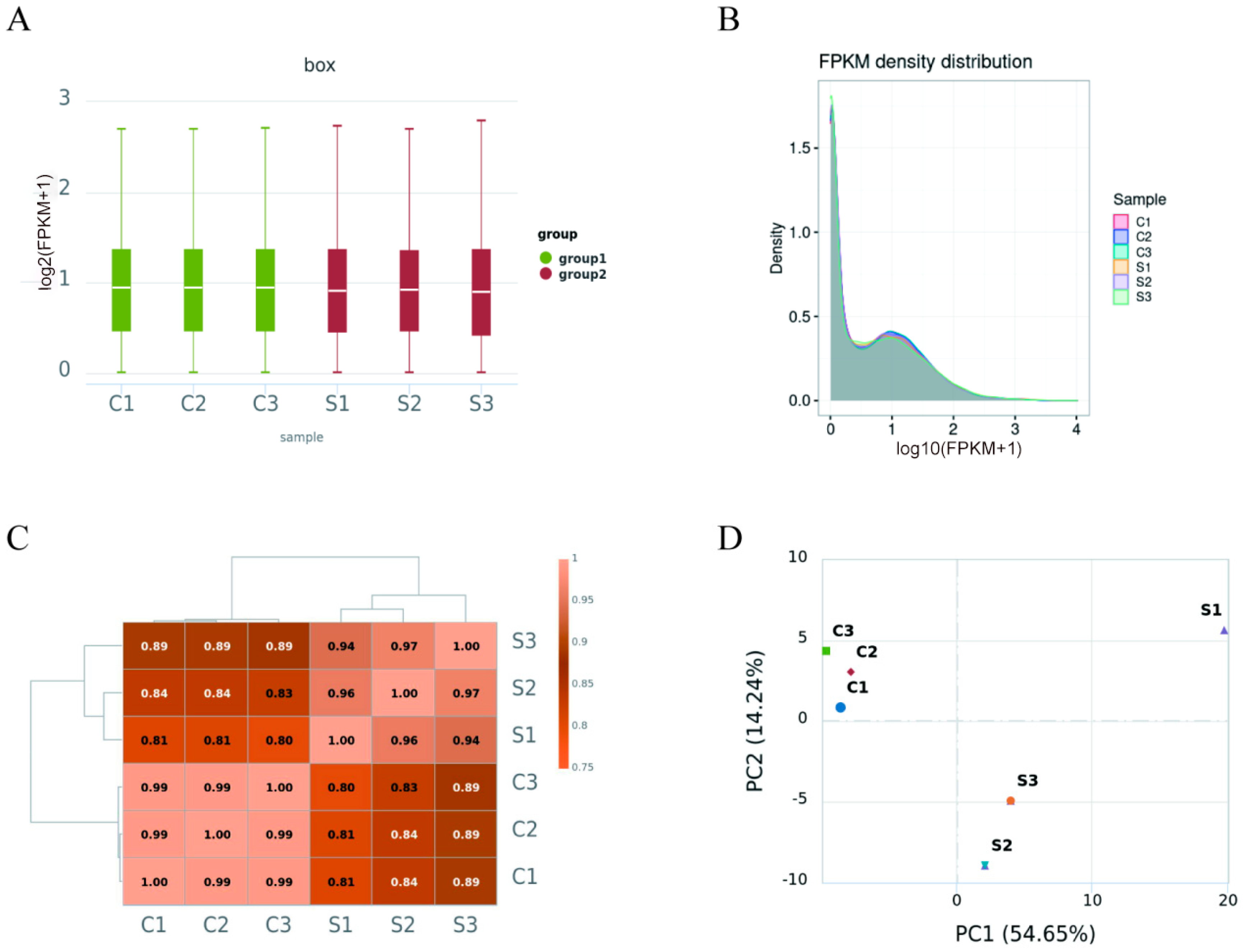
3.1. De Novo Transcriptome Assembly via Sequencing
3.2. Transcriptomic Mapping
3.3. Profile of Gene Expression
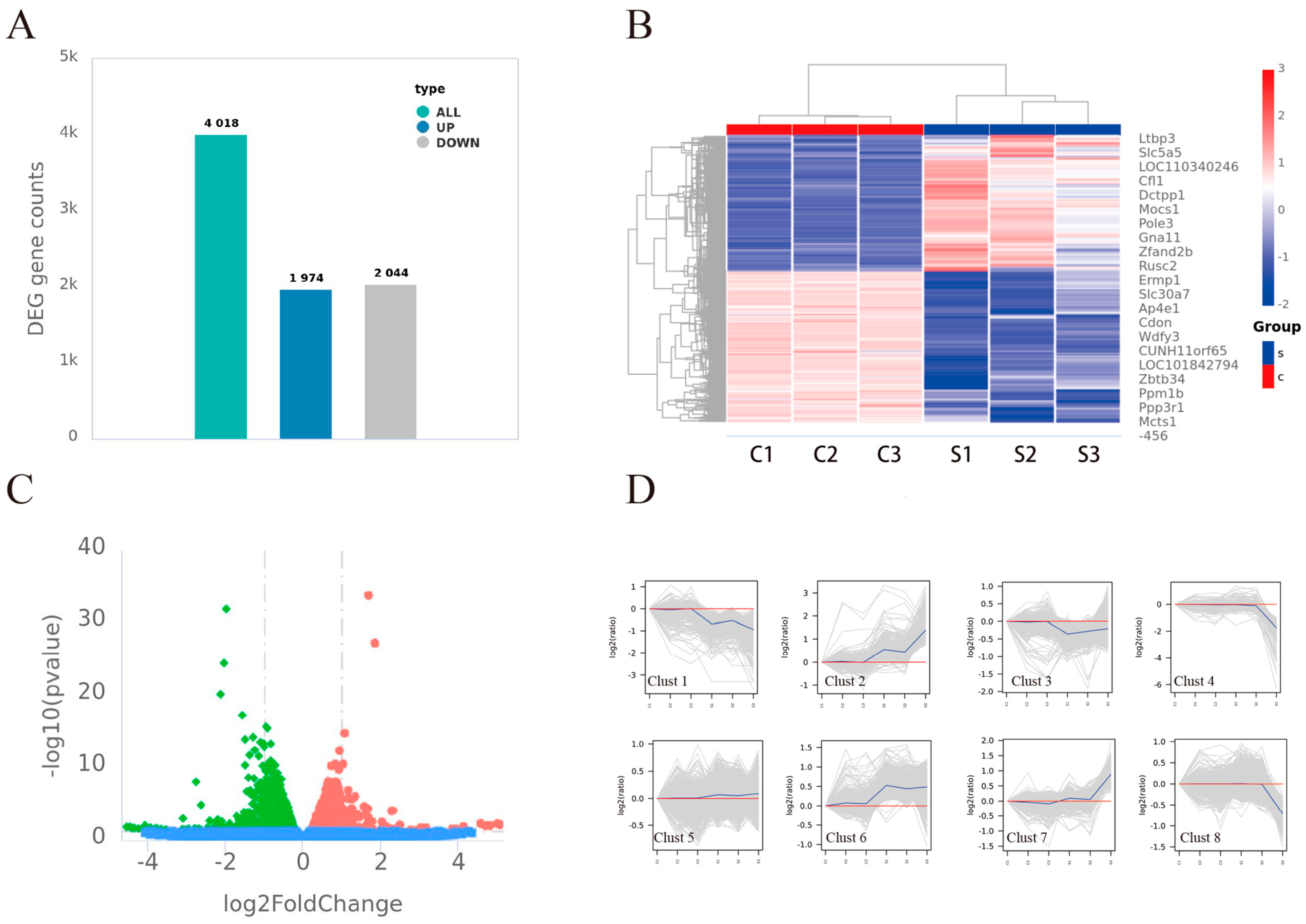
3.4. Analysis of Differential Expression
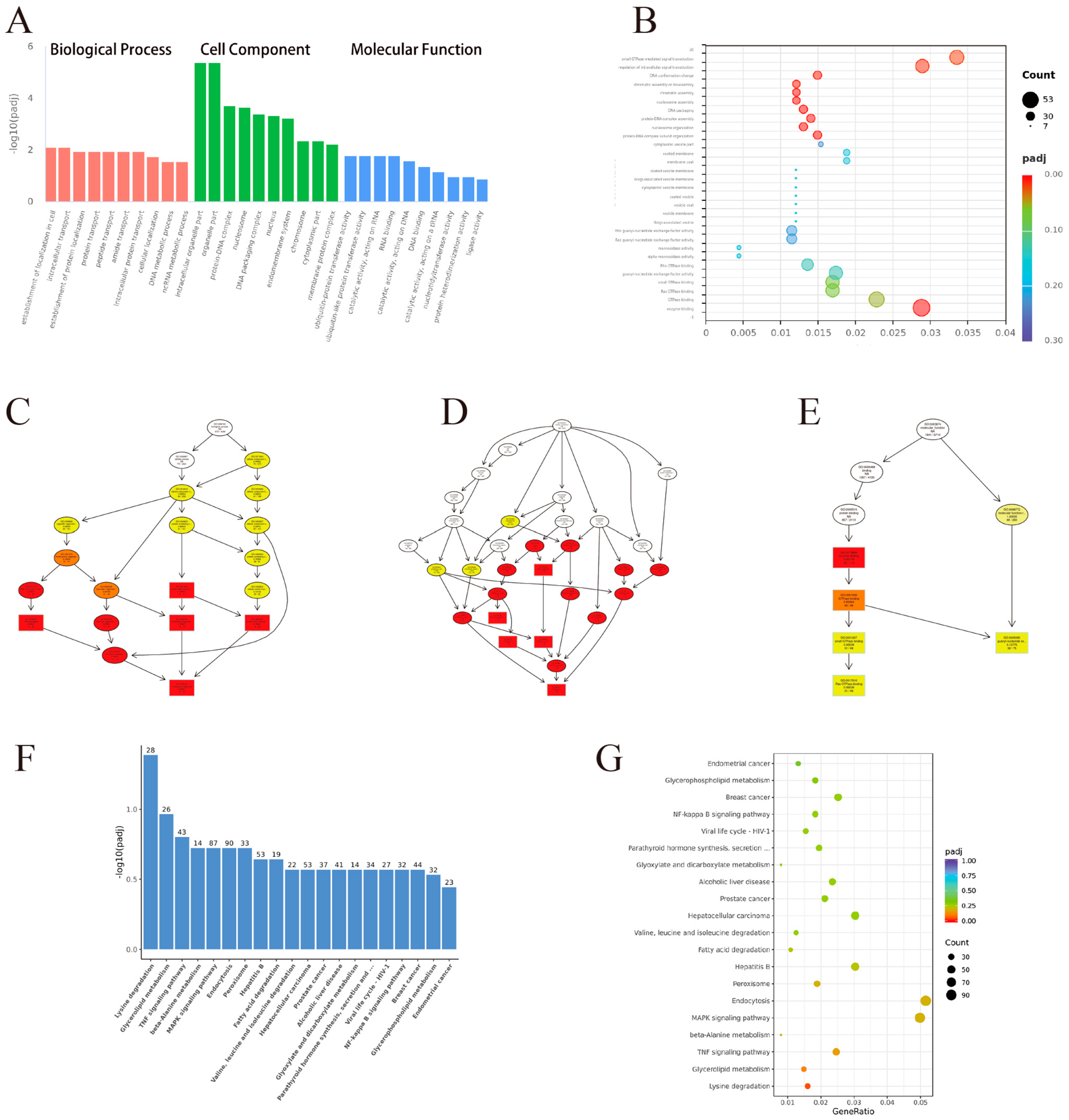
3.5. GO Enrichment Analysis
3.6. KEGG Enrichment Analysis
3.7. Additional RNA-seq Data Analysis
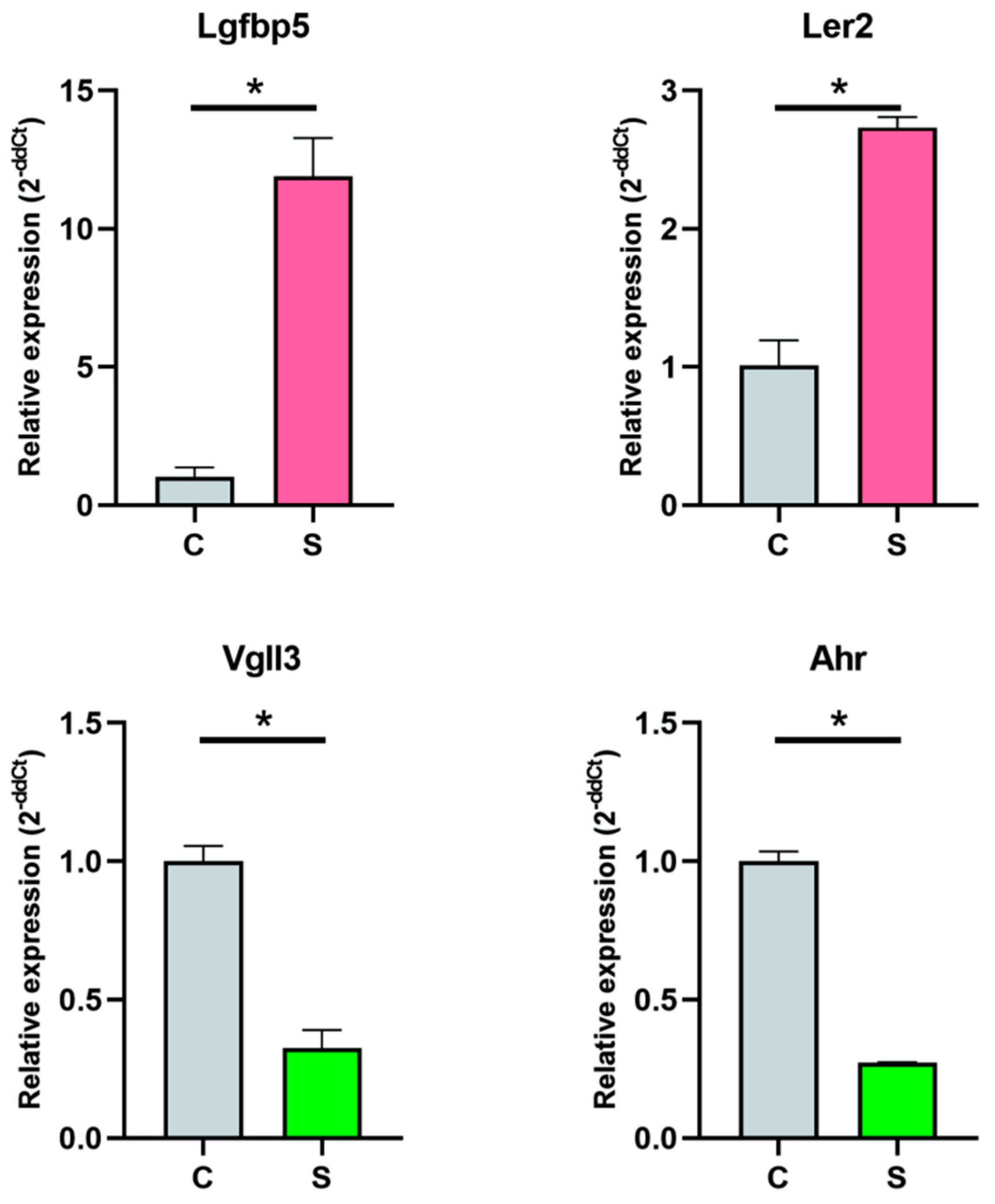
3.8. RT-qPCR Validation of Gene Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zewdie, G.; Akalu, M.; Tolossa, W.; Belay, H.; Deresse, G.; Zekarias, M.; Tesfaye, Y. A review of foot-and-mouth disease in Ethiopia: Epidemiological aspects, economic implications, and control strategies. Virol. J. 2023, 20, 299. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Abdullah, S.W.; Wu, J.; Tang, J.; Zhang, Y.; Dong, H.; Bai, M.; Wei, S.; Sun, S.; Guo, H. Foot-and-mouth disease virus downregulates vacuolar protein sorting 28 to promote viral replication. J. Virol. 2023, 97, e0018123. [Google Scholar] [CrossRef] [PubMed]
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Abdullah, S.W.; Li, P.; Wu, J.; Pei, C.; Mu, S.; Wang, Y.; Sun, S.; Guo, H. Heat Shock Protein 60 Is Involved in Viral Replication Complex Formation and Facilitates Foot and Mouth Virus Replication by Stabilizing Viral Nonstructural Proteins 3A and 2C. mBio 2022, 13, e0143422. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Li, Y.; Chu, H.; Jiang, Y.; Li, Z.; Wang, J.; Liu, F. Comparative transcriptomics analysis on Senecavirus A-infected and non-infected cells. Front. Vet. Sci. 2024, 11, 1431879. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Qu, M.; Li, Z.; Yin, X.; Tang, L.; Liu, X.; Sun, Y. Foot-and-mouth disease virus structural protein VP3 interacts with HDAC8 and promotes its autophagic degradation to facilitate viral replication. Autophagy 2023, 19, 2869–2883. [Google Scholar] [CrossRef]
- Weerawardhana, A.; Uddin, M.B.; Choi, J.H.; Pathinayake, P.; Shin, S.H.; Chathuranga, K.; Park, J.H.; Lee, J.S. Foot-and-mouth disease virus non-structural protein 2B downregulates the RLR signaling pathway via degradation of RIG-I and MDA5. Front. Immunol. 2022, 13, 1020262. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, K.; Luo, Z.; Nian, X.; Choudhury, S.K.M.; Zhu, Z.; Song, R.; Pei, J.; Huo, Y.; Li, Y.; et al. FMDV 3A Antagonizes the Effect of ANXA1 to Positively Modulate Viral Replication. J. Virol. 2022, 96, e0031722. [Google Scholar] [CrossRef]
- Westermann, A.J.; Vogel, J. Cross-species RNA-seq for deciphering host-microbe interactions. Nat. Rev. Genet. 2021, 22, 361–378. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Li, K.; Wang, C.; Yang, F.; Cao, W.; Zhu, Z.; Zheng, H. Virus-Host Interactions in Foot-and-Mouth Disease Virus Infection. Front. Immunol. 2021, 12, 571509. [Google Scholar] [CrossRef]
- Nie, Z.; Zhai, F.; Zhang, H.; Zheng, H.; Pei, J. The multiple roles of viral 3D(pol) protein in picornavirus infections. Virulence 2024, 15, 2333562. [Google Scholar] [CrossRef]
- Saeidian, A.H.; Youssefian, L.; Vahidnezhad, H.; Uitto, J. Research Techniques Made Simple: Whole-Transcriptome Sequencing by RNA-Seq for Diagnosis of Monogenic Disorders. J. Investig. Dermatol. 2020, 140, 1117–1126.e1111. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Huang, A.Y.; Wang, L.K.; Yoon, A.J.; Renteria, G.; Eskin, A.; Signer, R.H.; Dorrani, N.; Nieves-Rodriguez, S.; Wan, J.; et al. Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genet. Med. 2020, 22, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, J.; Zhang, B.; Wang, R.; Wang, C.; Li, X.; Liu, Y.; Zhang, Y. Transcriptome sequencing analysis of echovirus 30 infection reveals its potential pathogenesis. Front. Microbiol. 2022, 13, 958385. [Google Scholar] [CrossRef] [PubMed]
- Braspenning, S.E.; Verjans, G.; Mehraban, T.; Messaoudi, I.; Depledge, D.P.; Ouwendijk, W.J.D. The architecture of the simian varicella virus transcriptome. PLoS Pathog. 2021, 17, e1010084. [Google Scholar] [CrossRef]
- Embregts, C.W.E.; Wentzel, A.S.; den Dekker, A.T.; van Ijcken, W.F.; Stadhouders, R.; GeurtsvanKessel, C.H. Rabies virus uniquely reprograms the transcriptome of human monocyte-derived macrophages. Front. Cell. Infect. Microbiol. 2023, 13, 1013842. [Google Scholar] [CrossRef]
- Cheng, Y.; Sun, F.; Wang, L.; Gao, M.; Xie, Y.; Sun, Y.; Liu, H.; Yuan, Y.; Yi, W.; Huang, Z.; et al. Virus-induced p38 MAPK activation facilitates viral infection. Theranostics 2020, 10, 12223–12240. [Google Scholar] [CrossRef]
- DuShane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar] [CrossRef]
- Gonzalez Lopez Ledesma, M.M.; Costa Navarro, G.; Pallares, H.M.; Paletta, A.; De Maio, F.; Iglesias, N.G.; Gebhard, L.; Oviedo Rouco, S.; Ojeda, D.S.; de Borba, L.; et al. Dengue virus NS5 degrades ERC1 during infection to antagonize NF-kB activation. Proc. Natl. Acad. Sci. USA 2023, 120, e2220005120. [Google Scholar] [CrossRef]
- Albarnaz, J.D.; Ren, H.; Torres, A.A.; Shmeleva, E.V.; Melo, C.A.; Bannister, A.J.; Brember, M.P.; Chung, B.Y.; Smith, G.L. Molecular mimicry of NF-κB by vaccinia virus protein enables selective inhibition of antiviral responses. Nat. Microbiol. 2022, 7, 154–168. [Google Scholar] [CrossRef]
- Santoro, M.G.; Rossi, A.; Amici, C. NF-kappaB and virus infection: Who controls whom. Embo J. 2003, 22, 2552–2560. [Google Scholar] [CrossRef]
- Sumner, R.P.; Maluquer de Motes, C.; Veyer, D.L.; Smith, G.L. Vaccinia virus inhibits NF-κB-dependent gene expression downstream of p65 translocation. J. Virol. 2014, 88, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Liniger, M.; Gerber, M.; Renzullo, S.; García-Nicolás, O.; Ruggli, N. TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent. Viruses 2021, 13, 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, Y.; Chen, M. Viral strategies for triggering and manipulating mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef]
- van Riel, D.; Leijten, L.M.; van der Eerden, M.; Hoogsteden, H.C.; Boven, L.A.; Lambrecht, B.N.; Osterhaus, A.D.; Kuiken, T. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog. 2011, 7, e1002099. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Alkheraije, K.A. The prevalence of foot-and-mouth disease in Asia. Front. Vet. Sci. 2023, 10, 1201578. [Google Scholar] [CrossRef]
- Guo, M.; Xiong, M.; Peng, J.; Guan, T.; Su, H.; Huang, Y.; Yang, C.G.; Li, Y.; Boraschi, D.; Pillaiyar, T.; et al. Multi-omics for COVID-19: Driving development of therapeutics and vaccines. Natl. Sci. Rev. 2023, 10, nwad161. [Google Scholar] [CrossRef]
- Li, J.; Han, L.; Hao, Y.; Yuan, Y.; Wang, M.; Xin, X.; Wang, H.; Yu, F.; Zheng, C.; Shen, C. Comparative Transcriptome Analysis Reveals Different Host Cell Responses to Acute and Persistent Foot-and-Mouth Disease Virus Infection. Virol. Sin. 2020, 35, 52–63. [Google Scholar] [CrossRef]
- Zhao, F.R.; Xie, Y.L.; Liu, Z.Z.; Shao, J.J.; Li, S.F.; Zhang, Y.G.; Chang, H.Y. Transcriptomic analysis of porcine PBMCs in response to FMDV infection. Acta Trop. 2017, 173, 69–75. [Google Scholar] [CrossRef]
- Han, L.; Xin, X.; Wang, H.; Li, J.; Hao, Y.; Wang, M.; Zheng, C.; Shen, C. Cellular response to persistent foot-and-mouth disease virus infection is linked to specific types of alterations in the host cell transcriptome. Sci. Rep. 2018, 8, 5074. [Google Scholar] [CrossRef]
- Eschbaumer, M.; Stenfeldt, C.; Smoliga, G.R.; Pacheco, J.M.; Rodriguez, L.L.; Li, R.W.; Zhu, J.; Arzt, J. Transcriptomic Analysis of Persistent Infection with Foot-and-Mouth Disease Virus in Cattle Suggests Impairment of Apoptosis and Cell-Mediated Immunity in the Nasopharynx. PLoS ONE 2016, 11, e0162750. [Google Scholar] [CrossRef]
- Saravanan, S.; Guleria, N.; Ranjitha, H.B.; Sreenivasa, B.P.; Hosamani, M.; Prieto, C.; Umapathi, V.; Santosh, H.K.; Behera, S.; Dhanesh, V.V.; et al. Induction of antiviral and cell mediated immune responses significantly reduce viral load in an acute foot-and-mouth disease virus infection in cattle. Genomics 2021, 113, 4254–4266. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw_Reads | Raw_Bases | Error_Rate | Q20 | Q30 |
|---|---|---|---|---|---|
| C1 | 59608728 | 8.94 G | 0.01 | 99.23 | 97.75 |
| C2 | 48456016 | 7.27 G | 0.01 | 99.15 | 97.49 |
| C3 | 46872584 | 7.03 G | 0.01 | 99.18 | 97.57 |
| S1 | 51425968 | 7.71 G | 0.01 | 99.19 | 97.56 |
| S2 | 50985892 | 7.65 G | 0.01 | 99.19 | 97.58 |
| S3 | 51126662 | 7.67 G | 0.01 | 99.2 | 97.6 |
| Sample | Total_Reads | Total_Map | Unique_Map | Multi_Map |
|---|---|---|---|---|
| C1 | 57,923,034 | 53,335,422 (92.08%) | 51,050,372 (88.13%) | 2,285,050 (3.94%) |
| C2 | 47,517,542 | 44,024,204 (92.65%) | 42,258,755 (88.93%) | 1,765,449 (3.72%) |
| C3 | 45,965,232 | 42,241,759 (91.9%) | 40,624,971 (88.38%) | 1,616,788 (3.52%) |
| S1 | 50,429,350 | 39,210,770 (77.75%) | 37,623,031 (74.61%) | 1,587,739 (3.15%) |
| S2 | 49,797,192 | 40,176,416 (80.68%) | 38,469,646 (77.25%) | 1,706,770 (3.43%) |
| S3 | 49,814,424 | 40,082,756 (80.46%) | 38,523,751 (77.33%) | 1,559,005 (3.13%) |
| Sample | Exon | Intron | Intergenic |
|---|---|---|---|
| C1 | 7,285,406,051 (91.1852%) | 306,126,883 (3.8315%) | 398,150,357 (4.9833%) |
| C2 | 6,033,236,422 (91.4872%) | 241,780,946 (3.6663%) | 319,609,062 (4.8465%) |
| C3 | 5,823,037,349 (92.0338%) | 229,090,137 (3.6208%) | 274,940,524 (4.3455%) |
| S1 | 5,532,463,177 (94.1956%) | 125,114,966 (2.1302%) | 215,801,808 (3.6742%) |
| S2 | 5,586,065,790 (92.8252%) | 173,362,395 (2.8808%) | 258,400,739 (4.2939%) |
| S3 | 5,575,275,393 (92.8603%) | 177,995,772 (2.9646%) | 250,670,374 (4.1751%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Zheng, Z.; Liu, M.; Zhang, M.; Qu, X.; Ren, J.; Liao, M. Comparative Transcriptomics Analysis of Foot-and-Mouth Disease Virus-Infected Cell Model Systems. Vet. Sci. 2025, 12, 107. https://doi.org/10.3390/vetsci12020107
Ma H, Zheng Z, Liu M, Zhang M, Qu X, Ren J, Liao M. Comparative Transcriptomics Analysis of Foot-and-Mouth Disease Virus-Infected Cell Model Systems. Veterinary Sciences. 2025; 12(2):107. https://doi.org/10.3390/vetsci12020107
Chicago/Turabian StyleMa, Haibin, Zhenzhen Zheng, Min Liu, Mengsi Zhang, Xiaoyun Qu, Jingqiang Ren, and Ming Liao. 2025. "Comparative Transcriptomics Analysis of Foot-and-Mouth Disease Virus-Infected Cell Model Systems" Veterinary Sciences 12, no. 2: 107. https://doi.org/10.3390/vetsci12020107
APA StyleMa, H., Zheng, Z., Liu, M., Zhang, M., Qu, X., Ren, J., & Liao, M. (2025). Comparative Transcriptomics Analysis of Foot-and-Mouth Disease Virus-Infected Cell Model Systems. Veterinary Sciences, 12(2), 107. https://doi.org/10.3390/vetsci12020107