
Preparation and Antigenic Site Identification of Monoclonal Antibodies against PB1 Protein of H9N2 Subtype AIV



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus and Cells
2.2. Gene Amplification and Recombinant Expression of PB1
2.3. Mouse Immunization
2.4. Cell Fusion and Monoclonal Antibody (mAb) Preparation
2.5. ELISA Detection
2.6. IFA Experiment
2.7. Immunoblotting Analysis
2.8. Design of Truncated PB1 Gene and Identification of B Cell Epitope
2.9. Bioinformatics
3. Results
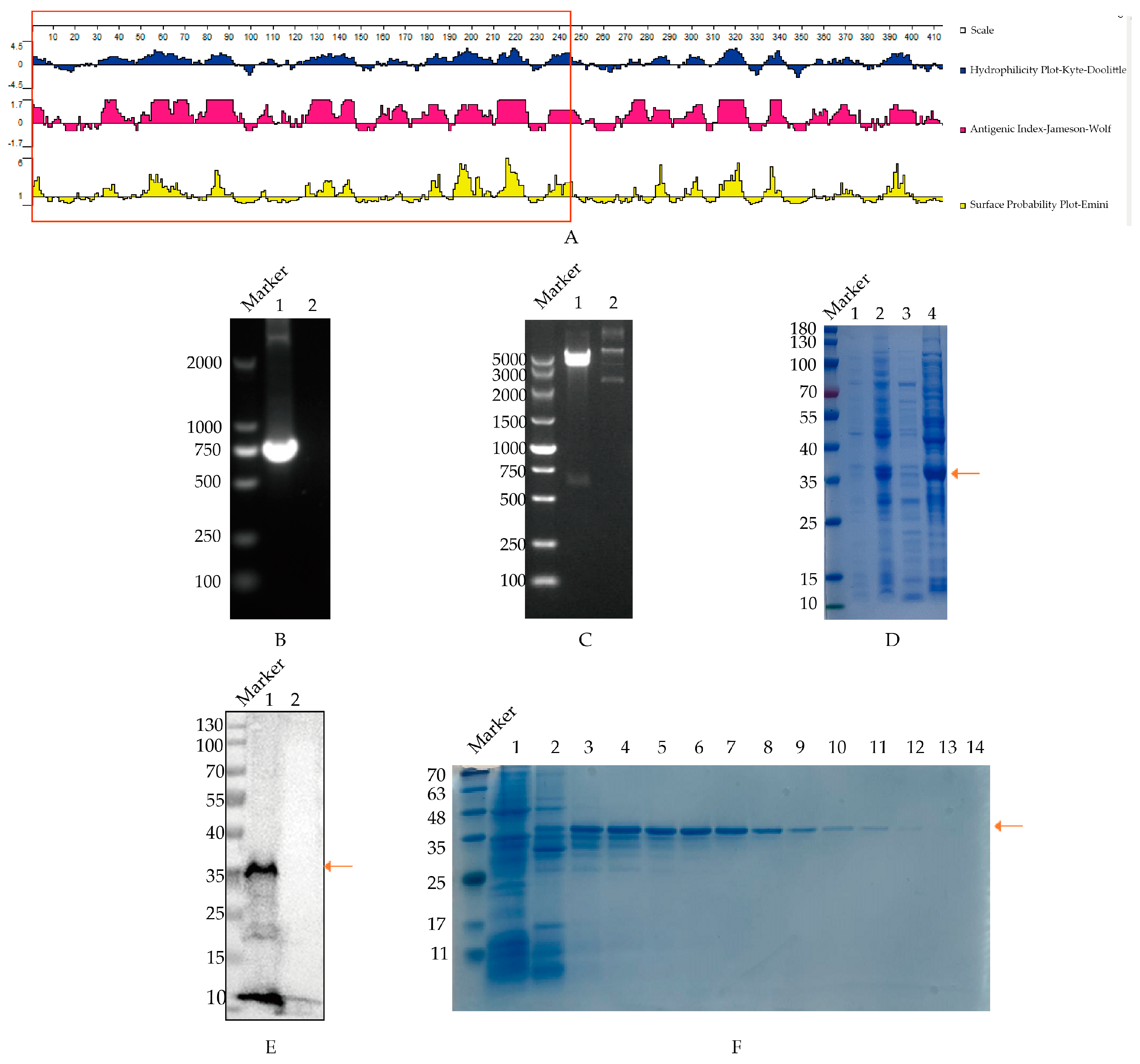
3.1. The Recombinant Construction and Expression of the PB1 Protein
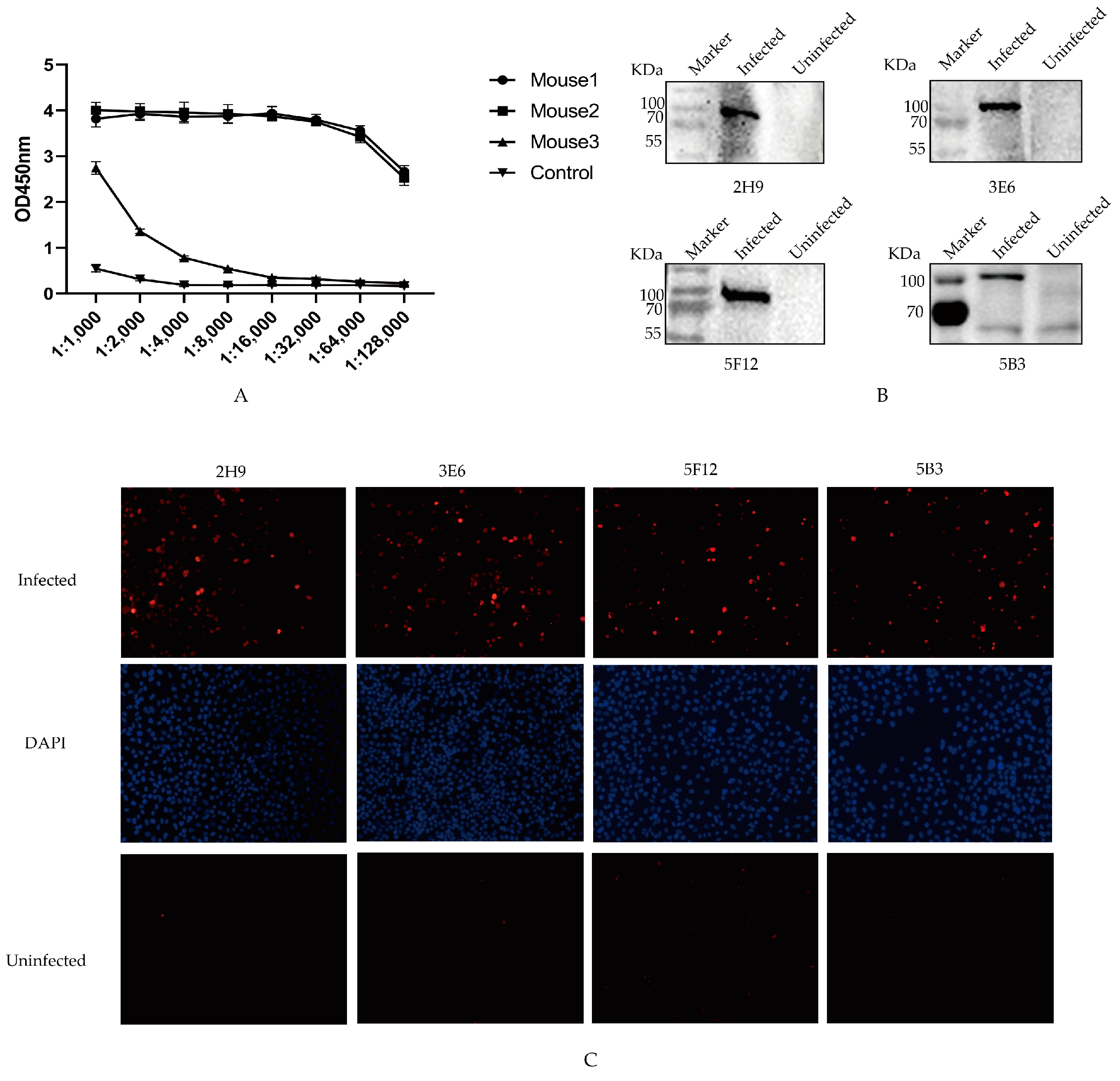
3.2. Screening of mAbs Specific to the PB1 Protein
3.3. B Cell Epitope Identification of mAbs Target to PB1 Protein
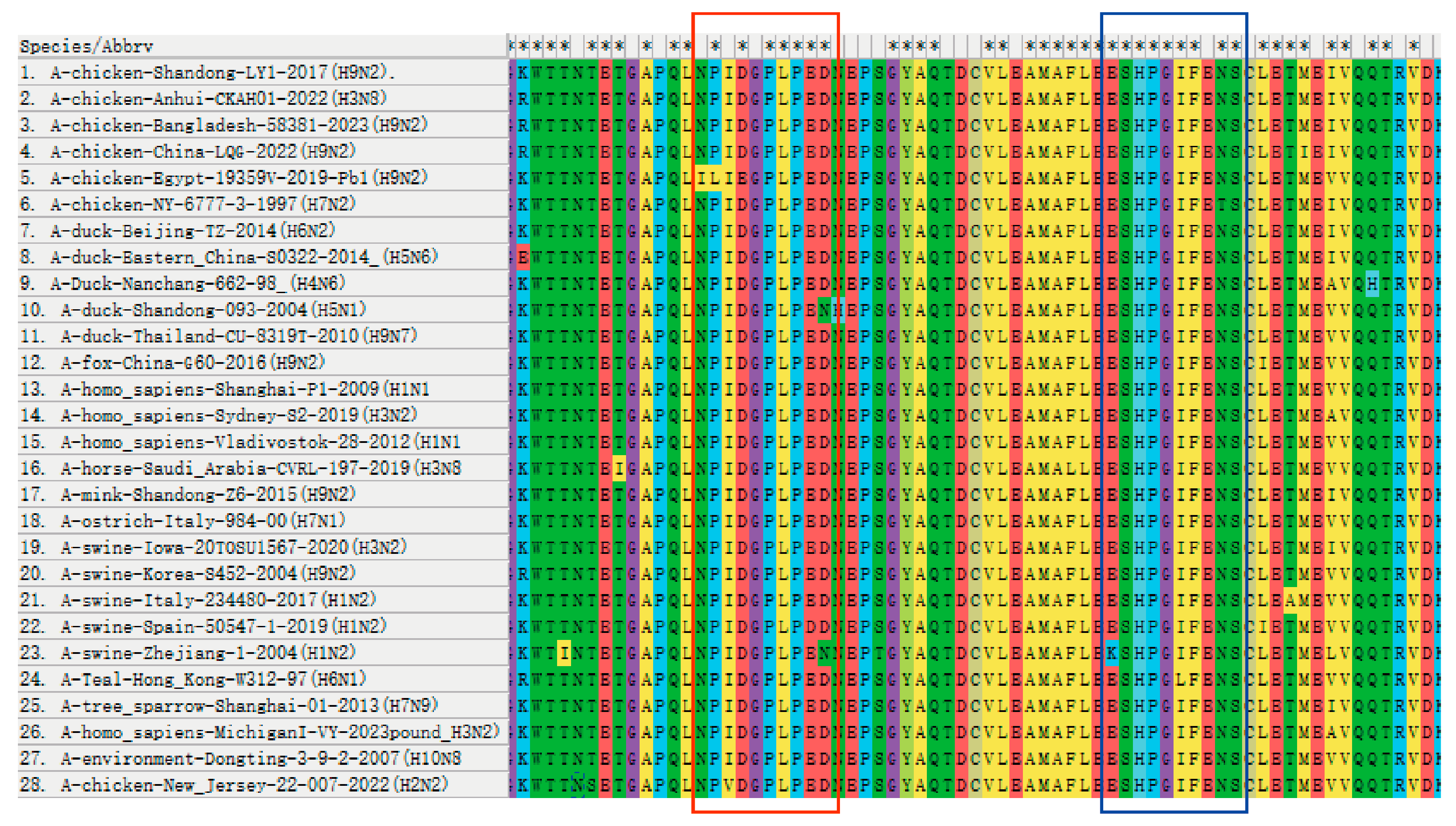
3.4. Conservative Identification of B Cell Epitopes across AIV Strains
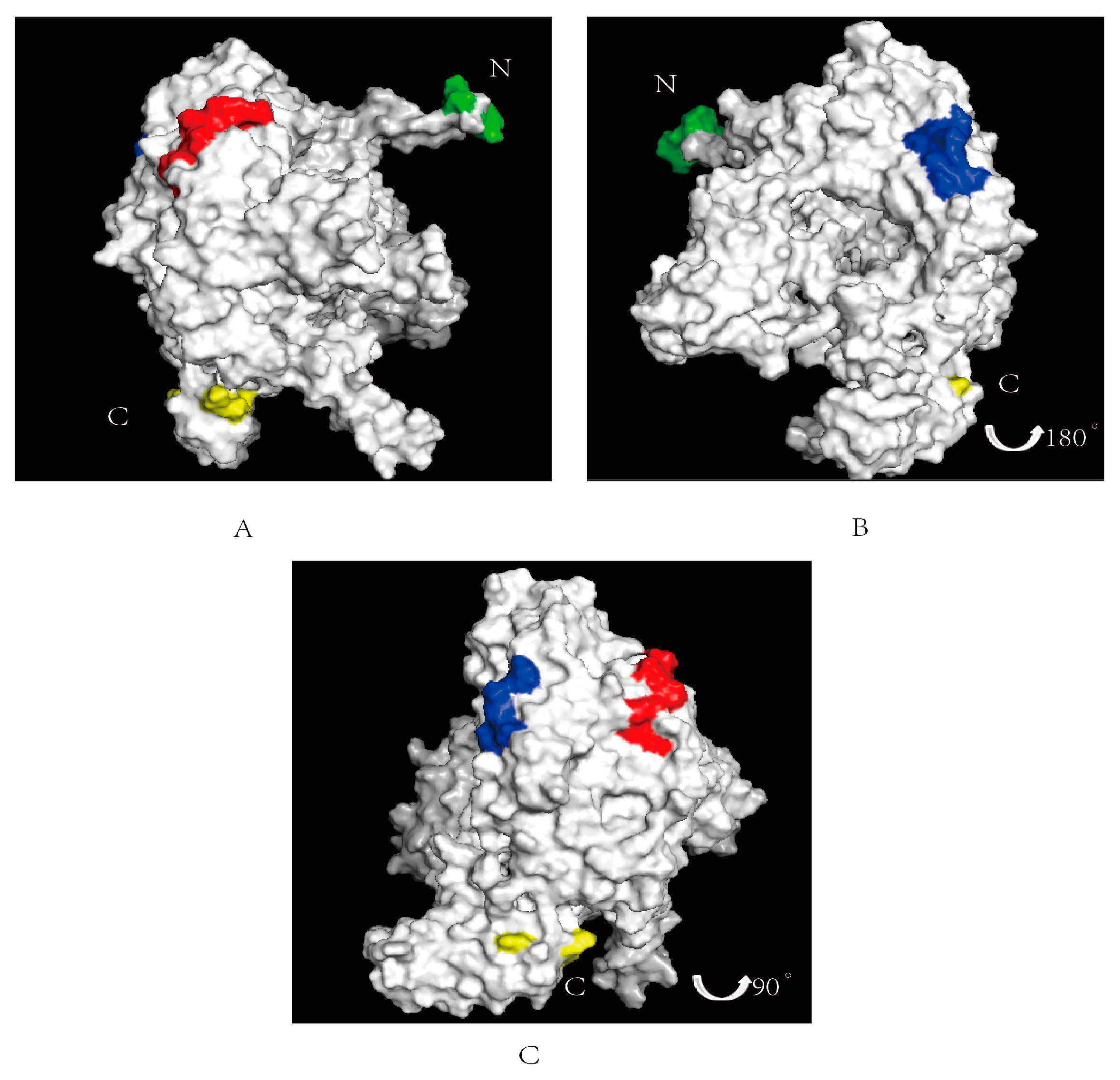
3.5. Spatial Structure Prediction of Epitopes in the PB1 Protein
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El Zowalaty, M.E.; Bustin, S.A.; Husseiny, M.I.; Ashour, H.M. Avian influenza: Virology, diagnosis and surveillance. Future Microbiol. 2013, 8, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Perdue, M.L.; Swayne, D.E. Public health risk from avian influenza viruses. Avian Dis. 2005, 49, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Homme, P.J.; Easterday, B.C. Avian influenza virus infections. I. Characteristics of influenza A-turkey-Wisconsin-1966 virus. Avian Dis. 1970, 14, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhao, L.; Guo, Y.; Zhao, Y.; Li, Y.; Chen, N.; Ping, J.J. Antigenic evolution characteristics and immunological evaluation of H9N2 avian influenza viruses from 1994–2019 in China. Viruses 2022, 14, 726. [Google Scholar] [CrossRef] [PubMed]
- Suarez, D.L. Avian influenza: Our current understanding. Anim. Health Res. Rev. 2010, 11, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Z.C.; Cao, Y.C. Host-Virus Interaction: How Host Cells Defend against Influenza A Virus Infection. Viruses 2020, 12, 376. [Google Scholar] [CrossRef] [PubMed]
- Suo, S.; Ren, X. Importance of interferon inducible trans-membrane proteins and retinoic acid inducible gene I for influenza virus replication: A review. Crit. Rev. Microbiol. 2016, 42, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kouba, T.; Drncova, P.; Cusack, S. Structural snapshots of actively transcribing influenza polymerase. Nat. Struct. Mol. Biol. 2019, 26, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, S.; Wang, B.; Ran, Y.; Tu, S.; Lin, Z.; Zhou, H. Proliferating cell nuclear antigen impairs the nuclear import of influenza A virus PB2 and suppresses virus replication. J. Med. Virol. 2023, 95, e28849. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.W.; Chen, G.W.; Sung, H.H.; Lin, R.J.; Yen, L.C.; Tseng, Y.L.; Liao, C.L. Naturally occurring mutations in PB1 affect influenza A virus replication fidelity, virulence, and adaptability. J. Biomed. Sci. 2019, 26, 55. [Google Scholar] [CrossRef] [PubMed]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, e141. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Pascua, P.N.; Lee, J.H.; Baek, Y.H.; Lee, O.J.; Kim, C.J.; Choi, Y.K. The polymerase acidic protein gene of influenza a virus contributes to pathogenicity in a mouse model. J. Virol. 2009, 83, 12325–12335. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lu, Y.; Liu, Q.; Zhou, Y.J. Inhibition of ongoing influenza A virus replication reveals different mechanisms of RIG-I activation. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Zhu, Q. The PB1 protein of influenza A virus inhibits the innate immune response by targeting MAVS for NBR1-mediated selective autophagic degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.N.; Zheng, Y.; Hao, S.S.; Zhang, Z.; Cai, J.X.; Zong, M.M.; Liu, Q.T. The molecular evolutionary characteristics of new isolated H9N2 AIV from East China and the function of vimentin on virus replication in MDCK cells. Virol. J. 2020, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Tan, D.; Shi, J.; Cui, P.; Jiang, Y.; Liu, L.; Chen, H. Complex reassortment of multiple subtypes of avian influenza viruses in domestic ducks at the Dongting Lake Region of China. J. Virol. 2013, 87, 9452–9462. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Serna Martin, I.; Karia, D.; Steyaert, J. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Suguitan, A.L., Jr.; Rivera, Y.; Simmons, C.P.; Lanzavecchia, A.; Sallusto, F.; Golding, H. Antigenic fingerprinting of H5N1 avian influenza using convalescent sera and monoclonal antibodies reveals potential vaccine and diagnostic targets. PLoS Med. 2009, 6, e1000049. [Google Scholar] [CrossRef] [PubMed]
- He, J.L.; Hsieh, M.S.; Chiu, Y.C.; Juang, R.H.; Wang, C.H. Preparation of monoclonal antibodies against poor immunogenic avian influenza virus proteins. J. Immunol. Methods 2013, 387, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.H.; Lee, T.T.; Chan, C.P.; Jin, D.Y. Influenza A virus PB1-F2 protein: An ambivalent innate immune modulator and virulence factor. J. Leukoc. Biol. 2020, 107, 763–771. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Name | Primer Sequence (5′–3′) | Fragment Length |
|---|---|---|---|
| PB1 | PB1-F | CGGGATCCATGGATGTCAATCCGACT | 735 bp |
| PB1-R | CCAAGCTTCCCGGGTGTTGCGATTGC | ||
| PB1-1 | PB1-1-F | CCGGAATTCATGGATGTCAATCCGACTTTACTTT | 492 bp |
| PB1-1-R | ACGCGTCGACTATCAGCCTTCCTGATTCATTGGCT | ||
| PB1-95 | PB1-95-F | CCGGAATTCCTTGAAGAATCTCACCCAGGGATCT | 453 bp |
| PB1-95-R | ACGCGTCGACCCCGGGTGTTGCGATTGCCCTCCTC | ||
| PB1-67 | PB1-67-F | CCGGAATTCAATCCGATTGATGGACCACTACCTG | 150 bp |
| PB1-67-R | ACGCGTCGACTTGCTGAACAATTTCCATCGTTTCA | ||
| PB1-17 | PB1-17-F | CCGGAATTCGCCATAAGTACCACATTCCCTTATA | 150 bp |
| PB1-17-R | ACGCGTCGACGAGTTGGGGTGCTCCAGTCTCTGTG | ||
| PB1-27 | PB1-27-F | CCGGAATTCGACCCTCCATACAGCCATGGAACAG | 150 bp |
| PB1-27-R | ACGCGTCGACGTCCTCAGGTAGTGGTCCATCAATC | ||
| PB1-37 | PB1-37-F | CCGGAATTCGGATACACCATGGACACAGTCAACA | 150 bp |
| PB1-37-R | ACGCGTCGACATCTGTTTGTGCATACCCACTCGGC | ||
| PB1-47 | PB1-47-F | CCGGAATTCCATCAATACTCAGAAAAGGGAAAGT | 150 bp |
| PB1-47-R | ACGCGTCGACTTCAAGGAAAGCCATTGCTTCCAAT | ||
| PB1-57 | PB1-57-F | CCGGAATTCACGAACACAGAGACTGGAGCACCCC | 150 bp |
| PB1-57-R | ACGCGTCGACCGAGTTTTCAAAGATCCCTGGGTGA |
| mAbs | Mouse Ascites Potency | Heavy Chain | Light Chain |
|---|---|---|---|
| 2H9 | 1:512,000 | IgG1 | Kappa |
| 3E6 | 1:1,024,000 | IgG1 | Kappa |
| 5B3 | 1:1,024,000 | IgG2a | Kappa |
| 5F12 | 1:1,024,000 | IgG2a | Kappa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, Y.; Yin, G.; Hu, J.; Liu, Y.; Huang, X.; Gao, Z.; Guo, X.; Jiang, T.; Sun, H.; Feng, X. Preparation and Antigenic Site Identification of Monoclonal Antibodies against PB1 Protein of H9N2 Subtype AIV. Vet. Sci. 2024, 11, 412. https://doi.org/10.3390/vetsci11090412
Cai Y, Yin G, Hu J, Liu Y, Huang X, Gao Z, Guo X, Jiang T, Sun H, Feng X. Preparation and Antigenic Site Identification of Monoclonal Antibodies against PB1 Protein of H9N2 Subtype AIV. Veterinary Sciences. 2024; 11(9):412. https://doi.org/10.3390/vetsci11090412
Chicago/Turabian StyleCai, Yiqin, Guihu Yin, Jianing Hu, Ye Liu, Xiangyu Huang, Zichen Gao, Xinyu Guo, Ting Jiang, Haifeng Sun, and Xiuli Feng. 2024. "Preparation and Antigenic Site Identification of Monoclonal Antibodies against PB1 Protein of H9N2 Subtype AIV" Veterinary Sciences 11, no. 9: 412. https://doi.org/10.3390/vetsci11090412
APA StyleCai, Y., Yin, G., Hu, J., Liu, Y., Huang, X., Gao, Z., Guo, X., Jiang, T., Sun, H., & Feng, X. (2024). Preparation and Antigenic Site Identification of Monoclonal Antibodies against PB1 Protein of H9N2 Subtype AIV. Veterinary Sciences, 11(9), 412. https://doi.org/10.3390/vetsci11090412