
Hypoxia-Mediated Long Non-Coding RNA Fragment Identified in Canine Oral Melanoma through Transcriptome Analysis

 , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Cell Lines and Cell Culture
2.3. Isolation of EVs
2.4. RNA Isolation from Cells, Plasma, and Plasma EVs
2.5. Next-Generation Sequencing
2.6. Bioinformatic Analysis
2.7. qPCR
2.8. Statistical Analysis
3. Results
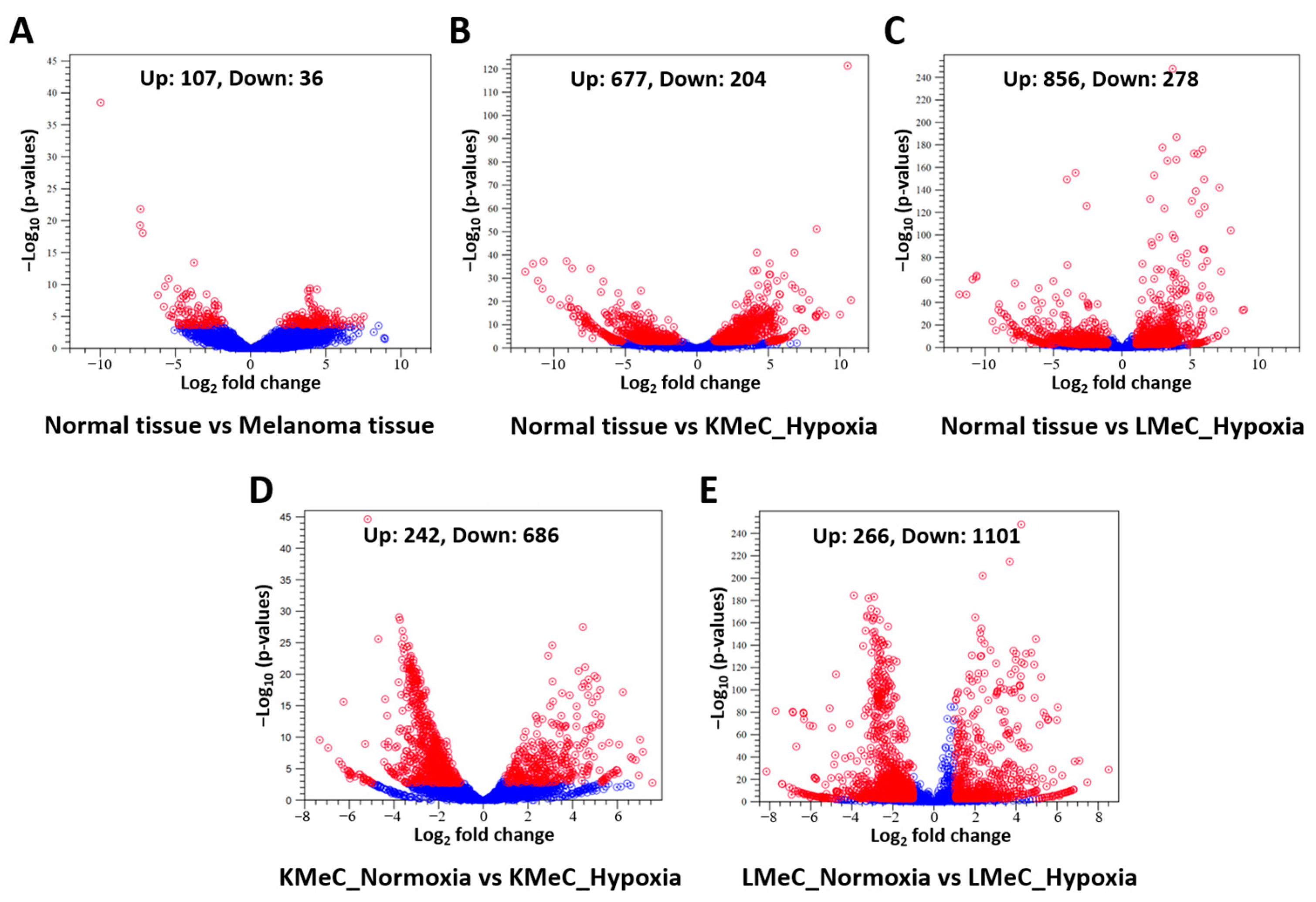
3.1. NGS Profiling of Hypoxia-Mediated ncRNAs
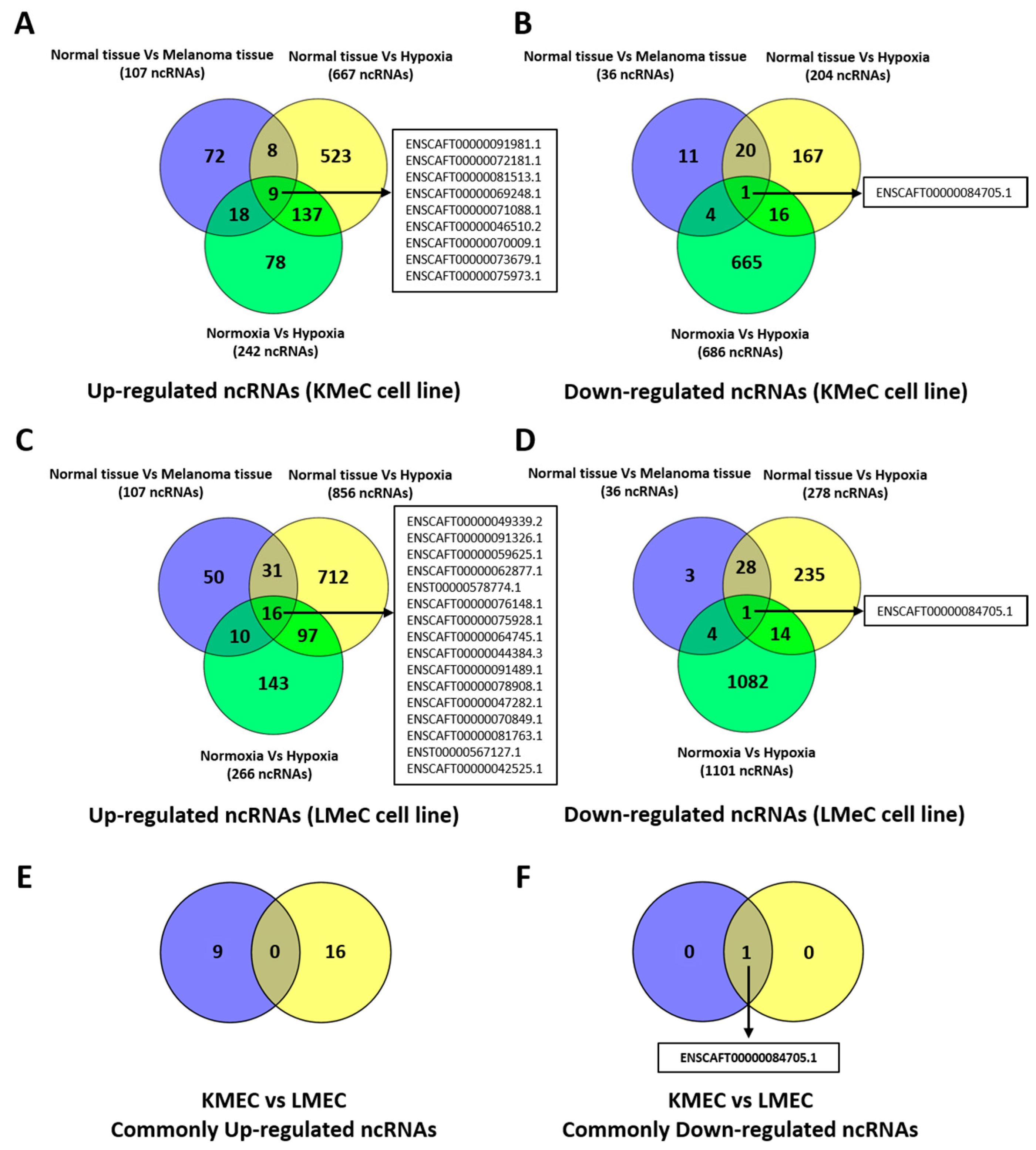
3.2. Identification of Target ncRNAs
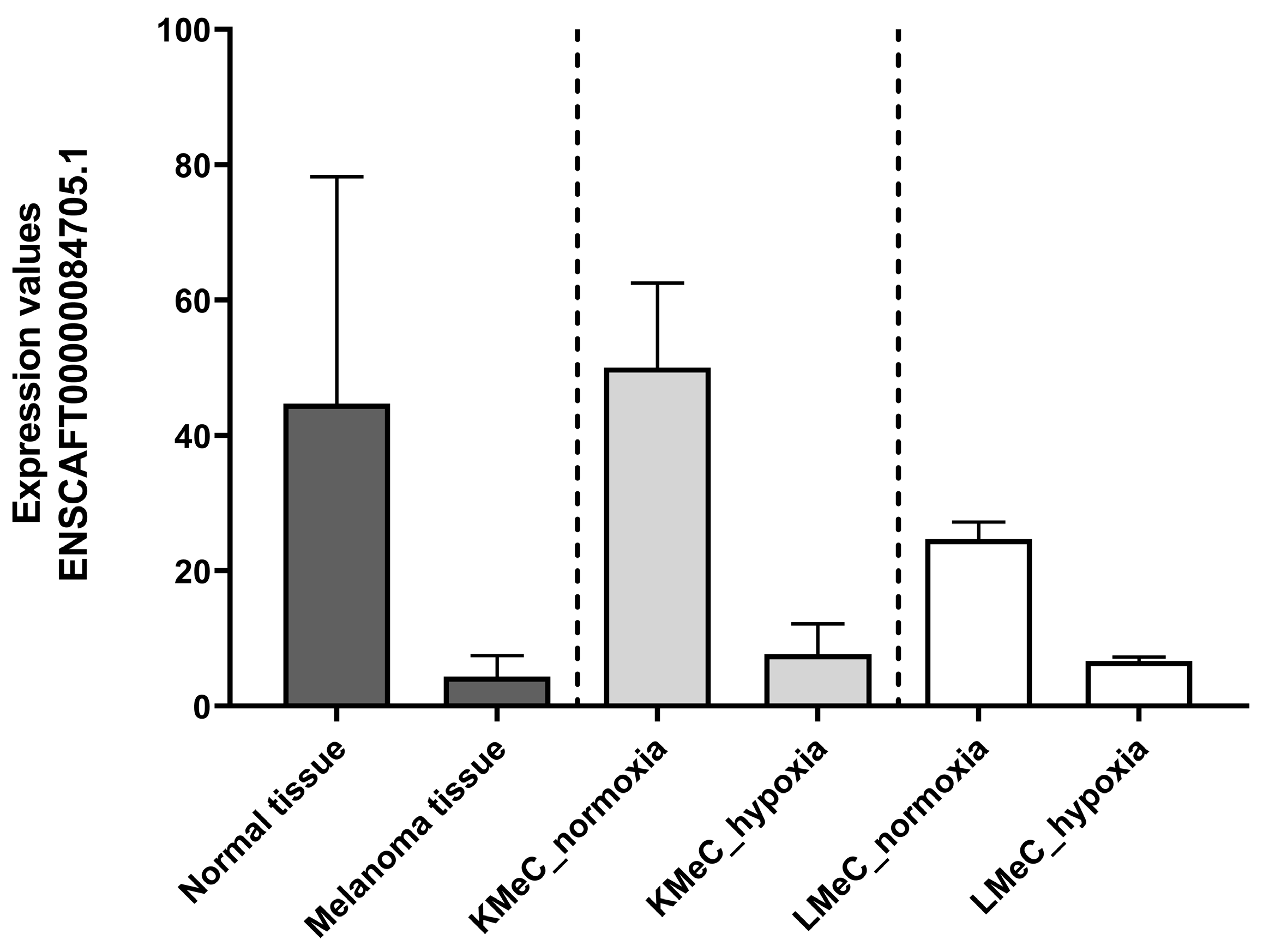
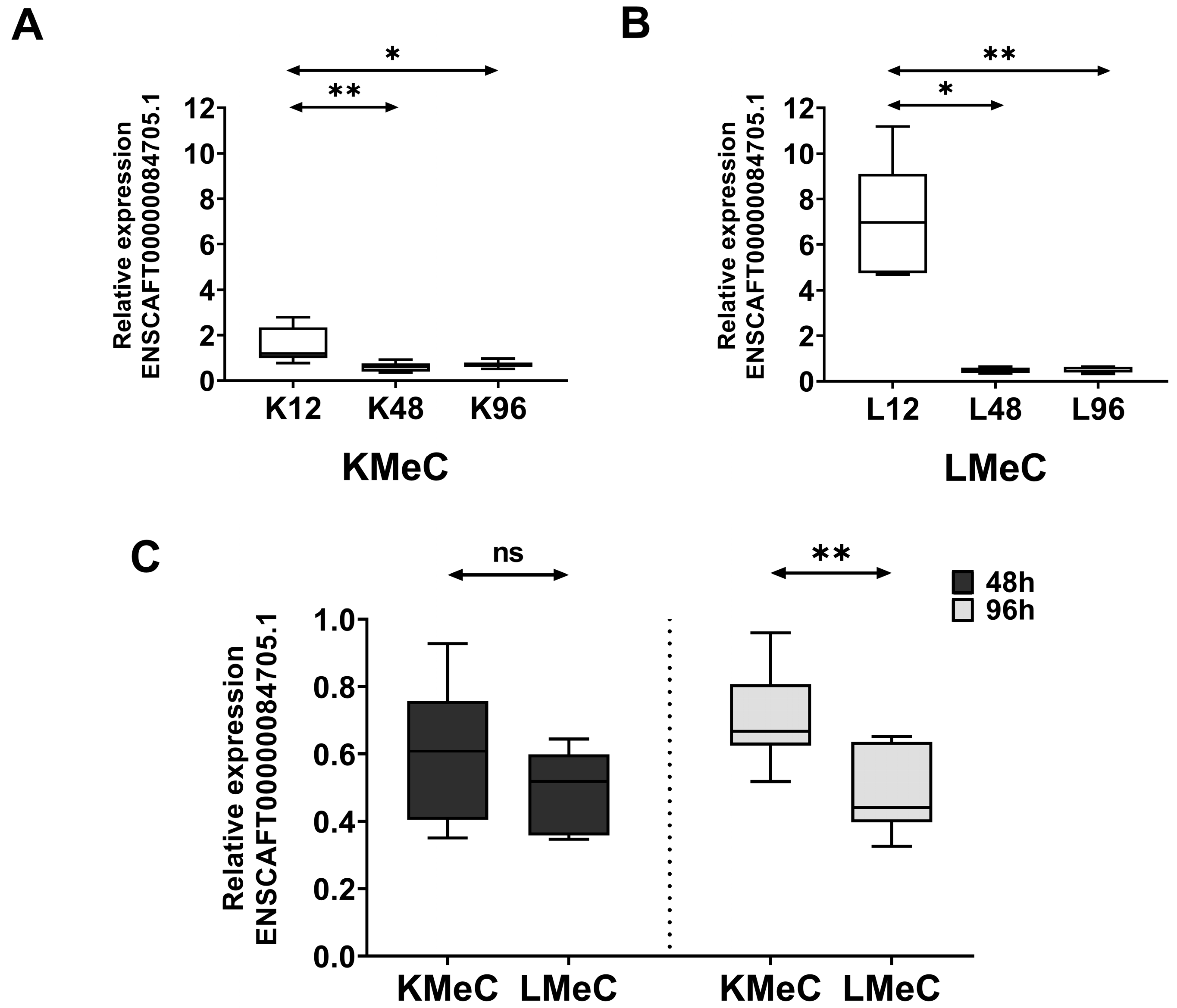
3.3. qRT-PCR Validation of Downregulated ncRNA (ENSCAFT00000084705.1)
3.3.1. Relative Expression in Hypoxic KMeC and LMeC Cells
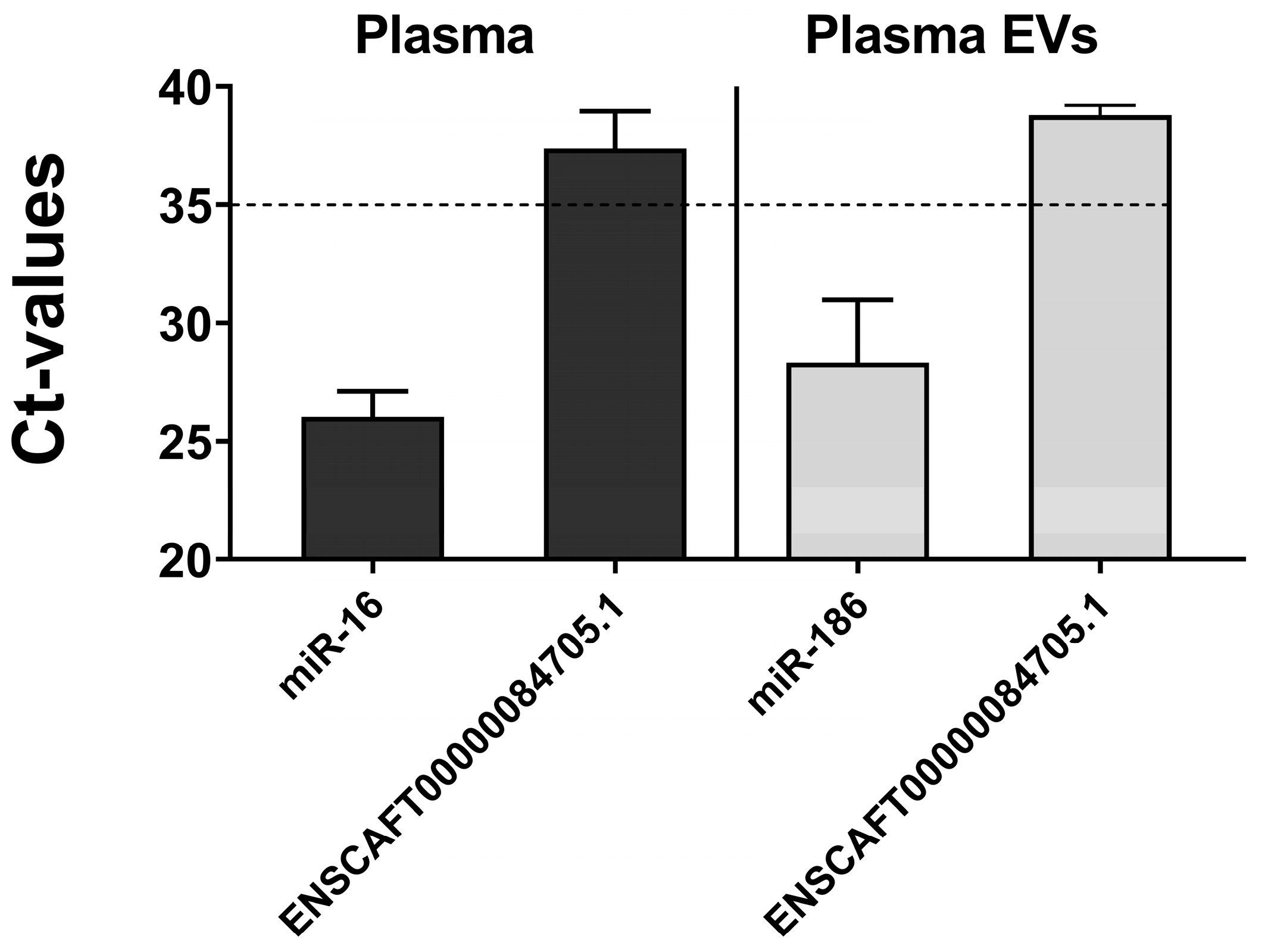
3.3.2. Relative Expression in Plasma and Plasma-Derived EVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally Occurring Melanomas in Dogs as Models for Non-UV Pathways of Human Melanomas. Pigment Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Prouteau, A.; André, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, L. A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma. Vet. Sci. 2021, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Mark Simpson, R. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.L.; Fenger, J.M.; London, C.A. Dogs as a Model for Cancer. Annu. Rev. Anim. Biosci. 2016, 4, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Van Der Weyden, L.; Patton, E.E.; Wood, G.A.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-Species Models of Human Melanoma. J. Pathol. 2016, 238, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Kato, D.; Nakagawa, T.; Arif, M.; Miura, N. Hypoxia-Related Y RNA Fragments as a Novel Potential Biomarker for Distinguishing Metastatic Oral Melanoma from Non-Metastatic Oral Melanoma in Dogs. Vet. Q. 2024, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Hino, Y.; Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.-W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Hypoxic miRNAs Expression are Different Between Primary and Metastatic Melanoma Cells. Gene 2021, 782, 145552. [Google Scholar] [CrossRef]
- Barreca, M.M.; Zichittella, C.; Alessandro, R.; Conigliaro, A. Hypoxia-induced Non-coding RNAs Controlling Cell Viability in Cancer. Int. J. Mol. Sci. 2021, 22, 1857. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of Hypoxia-Inducible Factor and Metabolic Pathways: Possible Targets of Cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia Inducible Factors in Cancer Stem Cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Hitte, C.; Le Béguec, C.; Cadieu, E.; Wucher, V.; Primot, A.; Prouteau, A.; Botherel, N.; Hédan, B.; Lindblad-Toh, K.; André, C.; et al. Genome-Wide Analysis of Long Non-Coding RNA Profiles in Canine Oral Melanomas. Genes 2019, 10, 477. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M. Tumor Hypoxia and Gene Expression—Implications for Malignant Progression and Therapy. Acta Oncol. 1998, 37, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Aberrantly Expressed snoRNA, snRNA, piRNA and tRFs in Canine Melanoma. Vet. Comp. Oncol. 2020, 18, 353–361. [Google Scholar] [CrossRef]
- Husna, A.A.; Rahman, M.M.; Lai, Y.C.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Identification of Melanoma-Specific Exosomal miRNAs as the Potential Biomarker for Canine Oral Melanoma. Pigment Cell Melanoma Res. 2021, 34, 1062–1073. [Google Scholar] [CrossRef]
- Husna, A.A.; Rahman, M.M.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Long Non-Coding RNA and Transfer RNA-Derived Small Fragments in Exosomes are Potential Biomarkers for Canine Oral Melanoma. Vet. Comp. Oncol. 2022, 20, 653–663. [Google Scholar] [CrossRef]
- Inoue, K.; Ohashi, E.; Kadosawa, T.; Hong, S.H.; Matsunaga, S.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Establishment and Characterization of Four Canine Melanoma Cell Lines. J. Vet. Med. Sci. 2004, 66, 1437–1440. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Lou, W.; Xiao, S.; Lin, K. Identification of a Hypoxia-suppressed lncRNA RAMP2-AS1 in Breast Cancer. Non-Coding RNA Res. 2024, 9, 782–795. [Google Scholar] [CrossRef]
- Kenneth, N.S.; Rocha, S. Regulation of Gene Expression by Hypoxia. Biochem. J. 2008, 414, 19–29. [Google Scholar] [CrossRef]
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression Under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef]
- Peng, X.; Gao, H.; Xu, R.; Wang, H.; Mei, J.; Liu, C. The Interplay between HIF-1α and Noncoding RNAs in Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 27. [Google Scholar] [CrossRef]
- Kim, W.S.; Vinayak, A.; Powers, B. Comparative Review of Malignant Melanoma and Histologically Well-Differentiated Melanocytic Neoplasm in the Oral Cavity of Dogs. Vet. Sci. 2021, 8, 261. [Google Scholar] [CrossRef]
- Gola, C.; Maniscalco, L.; Iussich, S.; Morello, E.; Olimpo, M.; Martignani, E.; Accornero, P.; Giacobino, D.; Mazzone, E.; Modesto, P.; et al. Hypoxia-associated Markers in the Prognosis of Oral Canine Melanoma. Vet. Pathol. 2024, 13, 03009858241244853. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Hypoxia and Hypoxia-Inducible Factors: Master Regulators of Metastasis. Clin. Cancer Res. 2010, 16, 5928–5935. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia Induced EMT: A Review on the Mechanism of Tumor Progression and Metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-Talk between HIF and p53 as Mediators of Molecular Responses to Physiological and Genotoxic Stresses. Mol. Cancer 2013, 12, 93. [Google Scholar] [CrossRef]
- Ibrahim, A.A.; Schmithals, C.; Kowarz, E.; Köberle, V.; Kakoschky, B.; Pleli, T.; Kollmar, O.; Nitsch, S.; Waidmann, O.; Finkelmeier, F.; et al. Hypoxia Causes Downregulation of Dicer in Hepatocellular Carcinoma, Which Is Required for Upregulation of Hypoxia-Inducible Factor 1α and Epithelial–Mesenchymal Transition. Clin. Cancer Res. 2017, 23, 3896–3905. [Google Scholar] [CrossRef]
- Cheng, J.C.; Klausen, C.; Leung, P.C.K. Hypoxia-Inducible Factor 1 Alpha Mediates Epidermal Growth Factor-Induced Down-regulation of E-Cadherin Expression and Cell Invasion in Human Ovarian Cancer Cells. Cancer Lett. 2013, 329, 197–206. [Google Scholar] [CrossRef]
- Kalluri, R. The Biology and Function of Exosomes in Cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L.; McIntyre, A. The Tumour Hypoxia Induced Non-Coding Transcriptome. Mol. Aspects Med. 2016, 47–48, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Coe, E.A.; Marine, J.C.; Vance, K.W. The Emerging Role of Long Non-Coding RNAs in Cutaneous Melanoma. Pigment Cell Melanoma Res. 2016, 29, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer: A Long Non-Coding RNA Point of View. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Age (Years) | Sex | Breed | Tumor Stage | Metastasis Status | Types of Samples |
|---|---|---|---|---|---|---|
| 1 | 13.3 | Male | M.D. | I | — | Tissue, Plasma |
| 2 | 10.3 | Male | Yorkshire | IV | — | Tissue, Plasma |
| 3 | 10.2 | Male | Chiwawa | IV | P | Tissue, Plasma |
| 4 | 12.7 | Male | M.D. | IV | P | Tissue, Plasma |
| 5 | 14.8 | Male | Mongrel | IV | P | Tissue, Plasma |
| 6 | 10 | Male | Golden Retriever | IV | — | Tissue, Plasma |
| 7 | 10.11 | Male | M.D. | I | — | Tissue |
| 8 | 7.11 | Male | M.D. | I | — | Tissue, Plasma |
| 9 | 10.9 | Male | M.D. | IV | — | Tissue, Plasma |
| 10 | 12 | Male | Shiba | IV | P | Tissue |
| 11 | 13 | Male | Pomerania | I | — | Tissue |
| 12 | 16.3 | Male | M.D. | IV | P | Tissue, Plasma |
| 13 | 11 | Male | M.D. | IV | P | Tissue, Plasma |
| 14 | 12 | Male | Mongrel | I | — | Tissue, Plasma |
| 15 | 11.1 | Male | M.D. | IV | P | Tissue, Plasma |
| 16 | 15.6 | Male | Pomeranian | II | P | Tissue, Plasma |
| 17 | 12.11 | Male | M.D. | IV | P | Tissue |
| 18 | 12.4 | Male | Shiba | IV | P | Tissue |
| 19 | 10.8 | Male | M.D. | IV | — | Tissue |
| 20 | 15.2 | Male | Shiba | I | — | Tissue |
| 21 | 12.4 | Female | M.D. | IV | — | Tissue, Plasma |
| 22 | 14.6 | Female | M.D. | II | P | Tissue |
| 23 | 15.2 | Female | Mongrel | IV | — | Tissue |
| 24 | 15.2 | Female | Mongrel | IV | P | Tissue |
| 25 | 8.2 | Female | M.D. | IV | P | Tissue, Plasma |
| 26 | 15.3 | Female | Mongrel | I | — | Tissue, Plasma |
| 27 | 15.3 | Female | Mongrel | I | — | Tissue, Plasma |
| 28 | 11.8 | Female | M.D. | I | — | Tissue, Plasma |
| 29 | 14 | Female | Dalmatian | II | P | Tissue, Plasma |
| 30 | 12.1 | Female | Toy poodle | IV | P | Tissue, Plasma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hino, Y.; Arif, M.; Rahman, M.M.; Husna, A.A.; Hasan, M.N.; Miura, N. Hypoxia-Mediated Long Non-Coding RNA Fragment Identified in Canine Oral Melanoma through Transcriptome Analysis. Vet. Sci. 2024, 11, 361. https://doi.org/10.3390/vetsci11080361
Hino Y, Arif M, Rahman MM, Husna AA, Hasan MN, Miura N. Hypoxia-Mediated Long Non-Coding RNA Fragment Identified in Canine Oral Melanoma through Transcriptome Analysis. Veterinary Sciences. 2024; 11(8):361. https://doi.org/10.3390/vetsci11080361
Chicago/Turabian StyleHino, Yasunori, Mohammad Arif, Md Mahfuzur Rahman, Al Asmaul Husna, MD Nazmul Hasan, and Naoki Miura. 2024. "Hypoxia-Mediated Long Non-Coding RNA Fragment Identified in Canine Oral Melanoma through Transcriptome Analysis" Veterinary Sciences 11, no. 8: 361. https://doi.org/10.3390/vetsci11080361
APA StyleHino, Y., Arif, M., Rahman, M. M., Husna, A. A., Hasan, M. N., & Miura, N. (2024). Hypoxia-Mediated Long Non-Coding RNA Fragment Identified in Canine Oral Melanoma through Transcriptome Analysis. Veterinary Sciences, 11(8), 361. https://doi.org/10.3390/vetsci11080361