
Integration Analysis of Transcriptome Sequencing and Whole-Genome Resequencing Reveal Wool Quality-Associated Key Genes in Zhexi Angora Rabbits
 , ,
, ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Sequencing
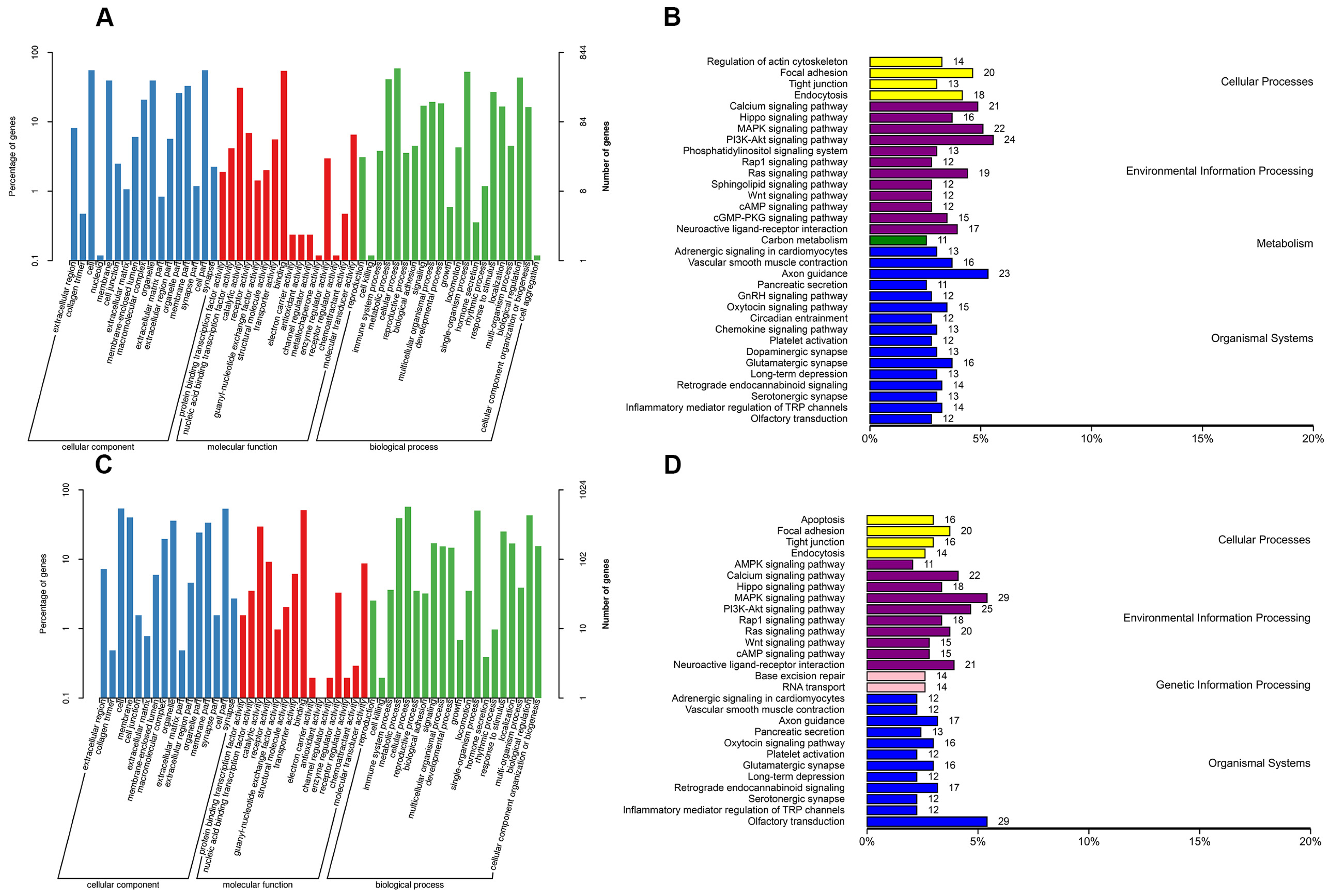
2.3. GO Enrichment and KEGG Pathway Analyses
2.4. Whole-Genome Resequencing
2.5. qPCR with Reverse Transcription
2.6. Joint Analysis of RNA-Seq and WGRS
2.7. Statistical Analysis
3. Results
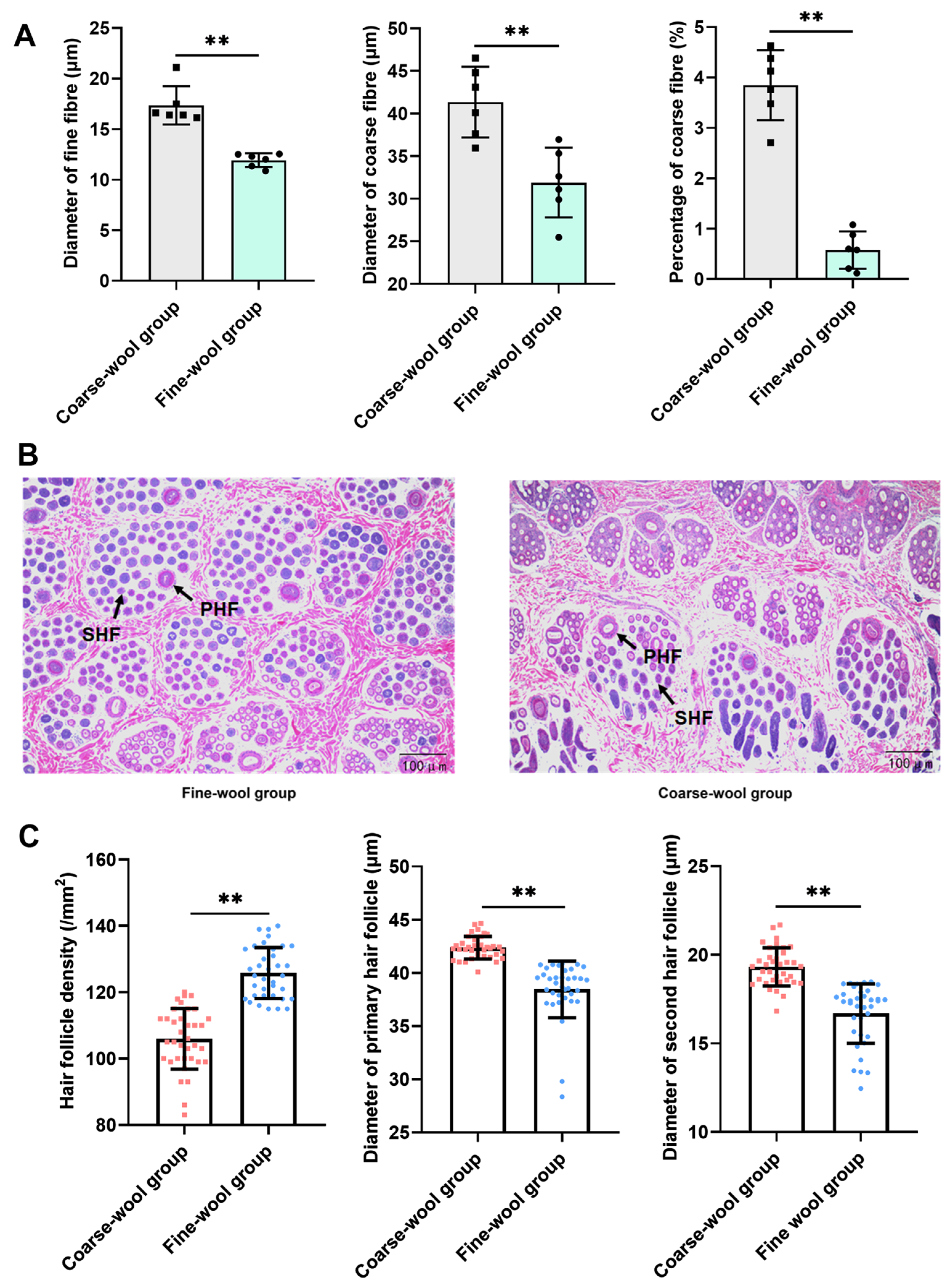
3.1. Morphological Characteristics of HFs in Zhexi Angora Rabbits
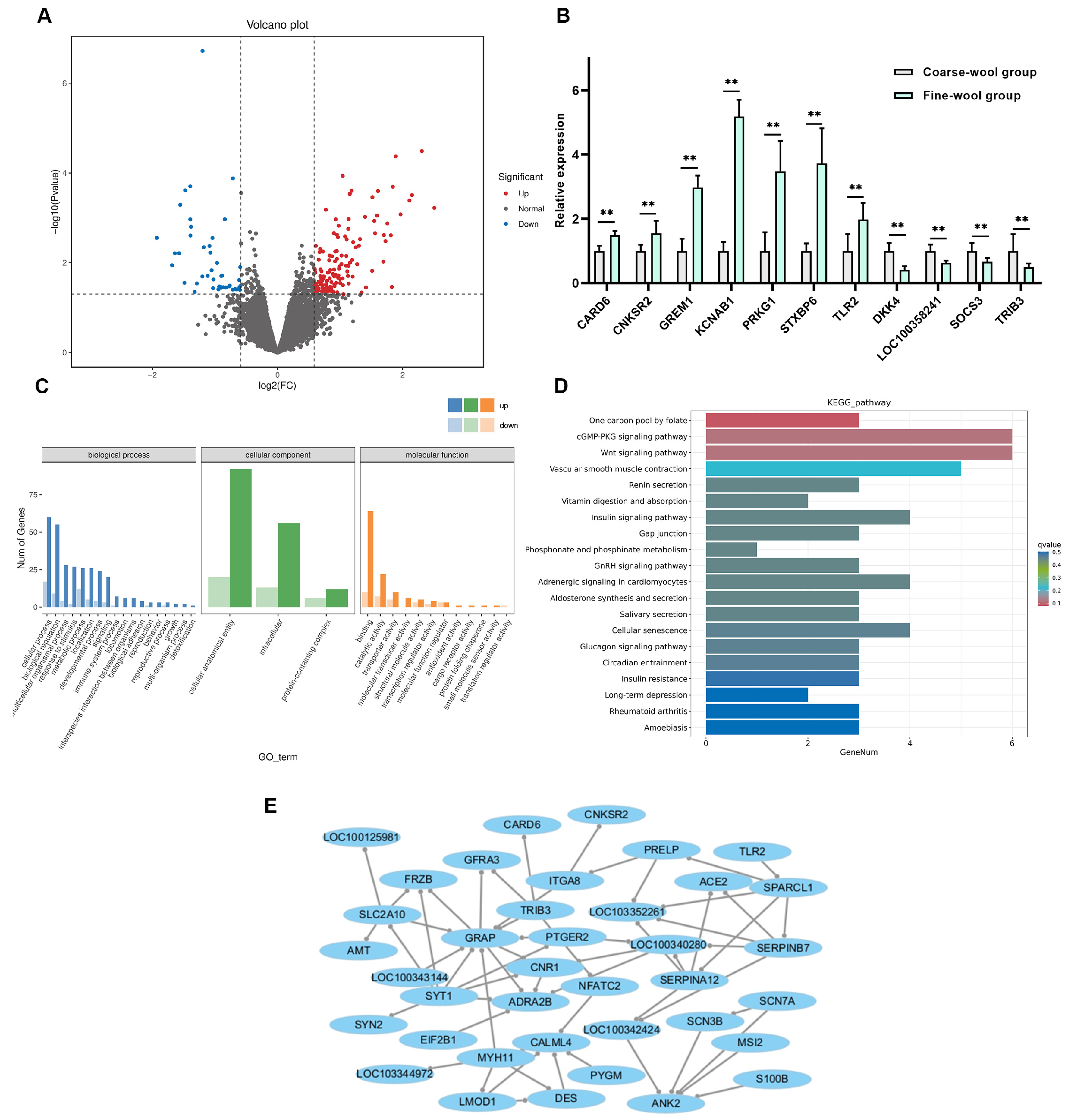
3.2. Screening of Fiber Formation-Associated DEGs Through RNA-Seq
3.3. Identification of Group-Specific Genetic Variation and Associated Genes Through WGRS
3.4. Joint Analysis of RNA-Seq and WGRS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The hair follicle as a dynamic miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, H. Fundamental hair follicle biology and fine fibre production in animals. Animal 2010, 4, 1490–1509. [Google Scholar] [CrossRef] [PubMed]
- Allafi, F.; Hossain, M.S.; Lalung, J.; Shaah, M.; Salehabadi, A.; Ahmad, M.I.; Shadi, A. Advancements in applications of natural wool fiber. J. Nat. Fibers 2022, 19, 497–512. [Google Scholar] [CrossRef]
- Laitala, K.; Klepp, I.G.; Henry, B. Does use matter? Comparison of environmental impacts of clothing based on fiber type. Sustainability 2018, 10, 2524. [Google Scholar] [CrossRef]
- Niranjan, S.; Sharma, S.; Gowane, G. Estimation of genetic parameters for wool traits in angora rabbit. Asian-Australas. J. Anim. Sci. 2011, 24, 1335–1340. [Google Scholar] [CrossRef]
- Doyle, E.K.; Preston, J.W.; McGregor, B.A.; Hynd, P.I. The science behind the wool industry. The importance and value of wool production from sheep. Anim. Front. 2021, 11, 15–23. [Google Scholar] [CrossRef]
- De Rochambeau, H.; Thébault, R.; Grun, J. Angora rabbit wool production: Non-genetic factors affecting quantity and quality of wool. Anim. Sci. 1991, 52, 383–393. [Google Scholar] [CrossRef]
- Murmu, S.B.; Debnath, S.; Bhutia, C.N. Evaluation of the german angora rabbit fiber produced in the northeast region of india. J. Nat. Fibers 2023, 20, 2210323. [Google Scholar] [CrossRef]
- Qian, Q.; Ma, J.; Zhang, G.; Xie, C.; Ren, L.; Qian, B. Breeding and Application of Zhexi Angora Rabbits. In Proceedings of the 11th World Rabbit Congress, Qingdao, China, 15–18 June 2016; pp. 861–864. [Google Scholar]
- Wang, D.; Xie, K.; Wang, Y.; Hu, J.; Li, W.; Yang, A.; Zhang, Q.; Ning, C.; Fan, X. Cost-effectively dissecting the genetic architecture of complex wool traits in rabbits by low-coverage sequencing. Genet. Sel. Evol. 2022, 54, 75. [Google Scholar] [CrossRef]
- Ning, C.; Xie, K.; Huang, J.; Di, Y.; Wang, Y.; Yang, A.; Hu, J.; Zhang, Q.; Wang, D.; Fan, X. Marker density and statistical model designs to increase accuracy of genomic selection for wool traits in angora rabbits. Front. Genet. 2022, 13, 968712. [Google Scholar] [CrossRef]
- Fatima, N.; Jia, L.; Liu, B.; Li, L.; Bai, L.; Wang, W.; Zhao, S.; Wang, R.; Liu, E. A homozygous missense mutation in the fibroblast growth factor 5 gene is associated with the long-hair trait in angora rabbits. BMC Genom. 2023, 24, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.; Hu, S.; Yang, N.; Wang, M.; Liu, M.; Li, J.; Xiao, Y.; Wu, X. Systematic analysis of non-coding rnas involved in the angora rabbit (Oryctolagus cuniculus) hair follicle cycle by rna sequencing. Front. Genet. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ding, H.; Wang, Y.; Cheng, G.; Wang, X.; Leng, T.; Zhao, H. Hair follicle transcriptome analysis reveals differentially expressed genes that regulate wool fiber diameter in angora rabbits. Biology 2023, 12, 445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.; Hao, Y.; Yang, N.; Wang, M.; Mei, M.; Wang, J.; Qiu, X.; Wu, X. Transcriptomic analysis reveals differentially expressed genes associated with wool length in rabbit. Anim. Genet. 2018, 49, 428–437. [Google Scholar] [CrossRef]
- Zhang, W.; Jin, M.; Li, T.; Lu, Z.; Wang, H.; Yuan, Z.; Wei, C. Whole-genome resequencing reveals selection signal related to sheep wool fineness. Animals 2023, 13, 2944. [Google Scholar] [CrossRef]
- Qi, Y.; Fu, S.; He, X.; Wang, B.; Da, L.; Te, R.; Yuejun, M.; Suzhen, S.; Zhang, W.; Liu, Y. Preliminary comparison of skin transcriptome from sheep with different wool fibre diameters. Anim. Prod. Sci. 2021, 61, 708–714. [Google Scholar] [CrossRef]
- Yue, L.; Lu, Z.; Guo, T.; Liu, J.; Yang, B.; Yuan, C. Key genes and metabolites that regulate wool fibre diameter identified by combined transcriptome and metabolome analysis. Genomics 2024, 116, 110886. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of samtools and bcftools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: Snps in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-freec: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K. Analyzing real-time pcr data by the comparative ct method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Li, S.; Chen, W.; Zheng, X.; Liu, Z.; Yang, G.; Hu, X.; Mou, C. Comparative investigation of coarse and fine wool sheep skin indicates the early regulators for skin and wool diversity. Genes 2020, 758, 144968. [Google Scholar] [CrossRef]
- Ding, H.; Zhao, H.; Cheng, G.; Yang, Y.; Wang, X.; Zhao, X.; Qi, Y.; Huang, D. Analyses of histological and transcriptome differences in the skin of short-hair and long-hair rabbits. BMC Genom. 2019, 20, 140. [Google Scholar] [CrossRef]
- Cui, C.-Y.; Kunisada, M.; Piao, Y.; Childress, V.; Ko, M.S.; Schlessinger, D. Dkk4 and eda regulate distinctive developmental mechanisms for subtypes of mouse hair. PLoS ONE 2010, 5, e10009. [Google Scholar] [CrossRef]
- Rendl, M.; Lewis, L.; Fuchs, E. Molecular dissection of mesenchymal–epithelial interactions in the hair follicle. PLoS Biol. 2005, 3, e331. [Google Scholar] [CrossRef]
- Chu, Q.; Cai, L.; Fu, Y.; Chen, X.; Yan, Z.; Lin, X.; Zhou, G.; Han, H.; Widelitz, R.B.; Chuong, C.-m. Dkk2/frzb in the dermal papillae regulates feather regeneration. Dev. Biol. 2014, 387, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Maglakelidze, N.; Gao, T.; Feehan, R.P.; Hobbs, R.P. Aire deficiency leads to the development of alopecia areata‒like lesions in mice. J. Investig. Dermatol. 2023, 143, 578–587.e573. [Google Scholar] [CrossRef] [PubMed]
- Niiyama, S.; Ishimatsu-Tsuji, Y.; Kishimoto, J. Niche formed by bone morphogenetic protein antagonists gremlin 1 and gremlin 2 in human hair follicles. Health Sci. Rep. 2022, 5, e486. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, Y.; Xing, Y.; Xu, W.; Guo, H.; Deng, F.; Ma, X.; Li, Y. Signaling, The balance of bmp6 and wnt10b regulates the telogen-anagen transition of hair follicles. Cell Commun. 2019, 17, 1–10. [Google Scholar]
- Xue, X.; Mi, Z.; Wang, Z.; Pang, Z.; Liu, H.; Zhang, F. High expression of ace2 on keratinocytes reveals skin as a potential target for SARS-CoV-2. J. Investig. Dermatol. 2021, 141, 206. [Google Scholar] [CrossRef]
- Selleri, S.; Arnaboldi, F.; Palazzo, M.; Gariboldi, S.; Zanobbio, L.; Opizzi, E.; Shirai, Y.; Balsari, A.; Rumio, C. Toll-like receptor agonists regulate β-defensin 2 release in hair follicle. Br. J. Dermatol. 2007, 156, 1172–1177. [Google Scholar] [CrossRef]
- Xiong, L.; Zhevlakova, I.; West, X.Z.; Gao, D.; Murtazina, R.; Horak, A.; Brown, J.M.; Molokotina, I.; Podrez, E.A.; Byzova, T.V. Tlr2 regulates hair follicle cycle and regeneration via bmp signaling. eLife 2024, 12, RP89335. [Google Scholar] [CrossRef]
- Xie, K.; Ning, C.; Yang, A.; Zhang, Q.; Wang, D.; Fan, X. Resequencing analyses revealed genetic diversity and selection signatures during rabbit breeding and improvement. Genes 2024, 15, 433. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, Y.; Yang, N.; Chen, Q.; Bao, Z.; Liu, M.; Hu, S.; Li, J.; Wu, X. Mir-218-5p regulates skin and hair follicle development through wnt/β-catenin signaling pathway by targeting sfrp2. J. Cell. Physiol. 2019, 234, 20329–20341. [Google Scholar] [CrossRef]
- Kim, B.-K.; Yoon, S.K. Expression of sfrp2 is increased in catagen of hair follicles and inhibits keratinocyte proliferation. Ann. Dermatol. 2014, 26, 79–87. [Google Scholar] [CrossRef]
- Bai, R.; Guo, Y.; Liu, W.; Song, Y.; Yu, Z.; Ma, X. The roles of wnt signaling pathways in skin development and mechanical-stretch-induced skin regeneration. Biomolecules 2023, 13, 1702. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, X.; Zhao, Q.; Stanic, V.; Lin, Z.; Yang, S.; Chen, T.; Chen, J.; Yang, Y. Hair loss caused by gain-of-function mutant trpv3 is associated with premature differentiation of follicular keratinocytes. J. Investig. Dermatol. 2021, 141, 1964–1974. [Google Scholar] [CrossRef]
- Shah, K.; Mehmood, S.; Jan, A.; Abbe, I.; Hussain Ali, R.; Khan, A.; Chishti, M.S.; Lee, K.; Ahmad, F.; Ansar, M. Sequence variants in nine different genes underlying rare skin disorders in 10 consanguineous families. Int. J. Dermatol. 2017, 56, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Li, A.G.; Liang, Y.-Y.; Owens, P.; He, W.; Lu, S.; Yoshimatsu, Y.; Wang, D.; Ten Dijke, P.; Lin, X. Smad7-induced β-catenin degradation alters epidermal appendage development. Dev. Cell 2006, 11, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S. Nod-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Zhang, L.; Wang, C.; Zhao, Z.; Zhang, M.; Li, X. Identification of microrna-21 target genes associated with hair follicle development in sheep. PeerJ 2019, 7, e7167. [Google Scholar] [CrossRef]
- Tian, D.; Pei, Q.; Jiang, H.; Guo, J.; Ma, X.; Han, B.; Li, X.; Zhao, K. Comprehensive analysis of the expression profiles of mrna, lncrna, circrna, and mirna in primary hair follicles of coarse sheep fetal skin. BMC Genom. 2024, 25, 574. [Google Scholar] [CrossRef]
- Tomcik, M.; Palumbo-Zerr, K.; Zerr, P.; Sumova, B.; Avouac, J.; Dees, C.; Distler, A.; Becvar, R.; Distler, O.; Schett, G. Tribbles homologue 3 stimulates canonical tgf-β signalling to regulate fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 609–616. [Google Scholar] [CrossRef]
- Grover, J.; Lee, E.; Mounkes, L.; Stewart, C.; Roughley, P. The consequence of prelp overexpression on skin. Matrix Biol. 2007, 26, 140–143. [Google Scholar] [CrossRef]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. Wnt signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Harel, S.; Higgins, C.A.; Cerise, J.E.; Dai, Z.; Chen, J.C.; Clynes, R.; Christiano, A.M. Pharmacologic inhibition of jak-stat signaling promotes hair growth. Sci. Adv. 2015, 1, e1500973. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gao, Y.; Fan, Z.; Xiao, X.; Chen, Y.; Si, Y.; Kong, D.; Wang, S.; Liao, M.; Chen, X. Amphiregulin promotes hair regeneration of skin-derived precursors via the pi3k and mapk pathways. Cell Prolif. 2021, 54, e13106. [Google Scholar] [CrossRef] [PubMed]
- Oshimori, N.; Fuchs, E. Paracrine tgf-β signaling counterbalances bmp-mediated repression in hair follicle stem cell activation. Cell Stem Cell 2012, 10, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, Z.; Wang, X.; Mo, M.; Zeng, S.B.; Xu, R.-H.; Wang, X.; Wu, Y. Pi3k/akt signaling pathway is essential for de novo hair follicle regeneration. Stem Cell Res. Ther. 2020, 11, 144. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | p-Value | log2 Fold Change | Chromosome | SNP Number | Indel Number |
|---|---|---|---|---|---|---|
| KCNAB1 | ENSOCUG00000007885 | 0.0001 | 1.0415 | 14 | 2 | |
| TRIB3 | ENSOCUG00000007636 | 0.0002 | −1.3960 | 4 | 7 | 1 |
| TLR2 | ENSOCUG00000003367 | 0.0010 | 1.3993 | 15 | 2 | |
| STXBP6 | ENSOCUG00000005144 | 0.0015 | 0.9784 | 17 | 1 | |
| PRKG1 | ENSOCUG00000011473 | 0.0058 | 0.6725 | 18 | 1238 | 184 |
| ALDH1L2 | ENSOCUG00000006962 | 0.0060 | −1.0734 | 4 | 1 | |
| CNKSR2 | ENSOCUG00000014660 | 0.0089 | 0.8258 | X | 1 | |
| PRELP | ENSOCUG00000009707 | 0.0141 | 0.6867 | 16 | 2 | |
| CARD6 | ENSOCUG00000005374 | 0.0232 | 1.0014 | 11 | 1 | 2 |
| ANK2 | ENSOCUG00000010618 | 0.0265 | 0.6598 | 15 | 1 | |
| CDH19 | ENSOCUG00000017601 | 0.0461 | 1.3454 | 9 | 1 | |
| CA10 | ENSOCUG00000002345 | 0.0495 | 0.6092 | 19 | 1 |
| Overlapping Signaling Pathways | RNA-Seq | WGRS |
|---|---|---|
| Wnt signaling pathway | DKK4, FMNL1, FRZB, NFATC2, PLCB4, RSPO1 | INVS, CSNK1A1, RSPO2, DAAM2, PRKCA, TCF7L2 |
| JAK-STAT signaling pathway | FHL1, SOCS3 | STAT4, BCL2L1, STAT5B, SOCS2, IL13RA1, SOCS6, PTPN11, SOCS7, AKT3 |
| MAPK signaling pathway | GRAP, LOC100358241 | PLA2G4E, ANGPT1, PDGFRL, PPM1B, PRKCA, WDR7 |
| TGF-β signaling pathway | GREM1 | TGIF2, BMP6, SMAD5, RBL1, LTBP1 |
| PI3K-Akt signaling pathway | ITGA8, LOC100353654, TLR2 | PPP2R3A, COL6A6, BCL2L1, CREB5, COL4A3, TLR2, ANGPT1, PRKAA2 |
| VEGF signaling pathway | NFATC2 | PLA2G4E, PRKCA, PLA2G4F, PPP3CA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B.; Yu, Y.; Sun, S.; Cai, J.; Bao, Z.; Chen, Y.; Wu, X. Integration Analysis of Transcriptome Sequencing and Whole-Genome Resequencing Reveal Wool Quality-Associated Key Genes in Zhexi Angora Rabbits. Vet. Sci. 2024, 11, 651. https://doi.org/10.3390/vetsci11120651
Zhao B, Yu Y, Sun S, Cai J, Bao Z, Chen Y, Wu X. Integration Analysis of Transcriptome Sequencing and Whole-Genome Resequencing Reveal Wool Quality-Associated Key Genes in Zhexi Angora Rabbits. Veterinary Sciences. 2024; 11(12):651. https://doi.org/10.3390/vetsci11120651
Chicago/Turabian StyleZhao, Bohao, Yongqi Yu, Shaoning Sun, Jiawei Cai, Zhiyuan Bao, Yang Chen, and Xinsheng Wu. 2024. "Integration Analysis of Transcriptome Sequencing and Whole-Genome Resequencing Reveal Wool Quality-Associated Key Genes in Zhexi Angora Rabbits" Veterinary Sciences 11, no. 12: 651. https://doi.org/10.3390/vetsci11120651
APA StyleZhao, B., Yu, Y., Sun, S., Cai, J., Bao, Z., Chen, Y., & Wu, X. (2024). Integration Analysis of Transcriptome Sequencing and Whole-Genome Resequencing Reveal Wool Quality-Associated Key Genes in Zhexi Angora Rabbits. Veterinary Sciences, 11(12), 651. https://doi.org/10.3390/vetsci11120651