
Mechanistic Insights into Influenza A Virus-Induced Cell Death and Emerging Treatment Strategies


Simple Summary
Abstract
1. Introduction
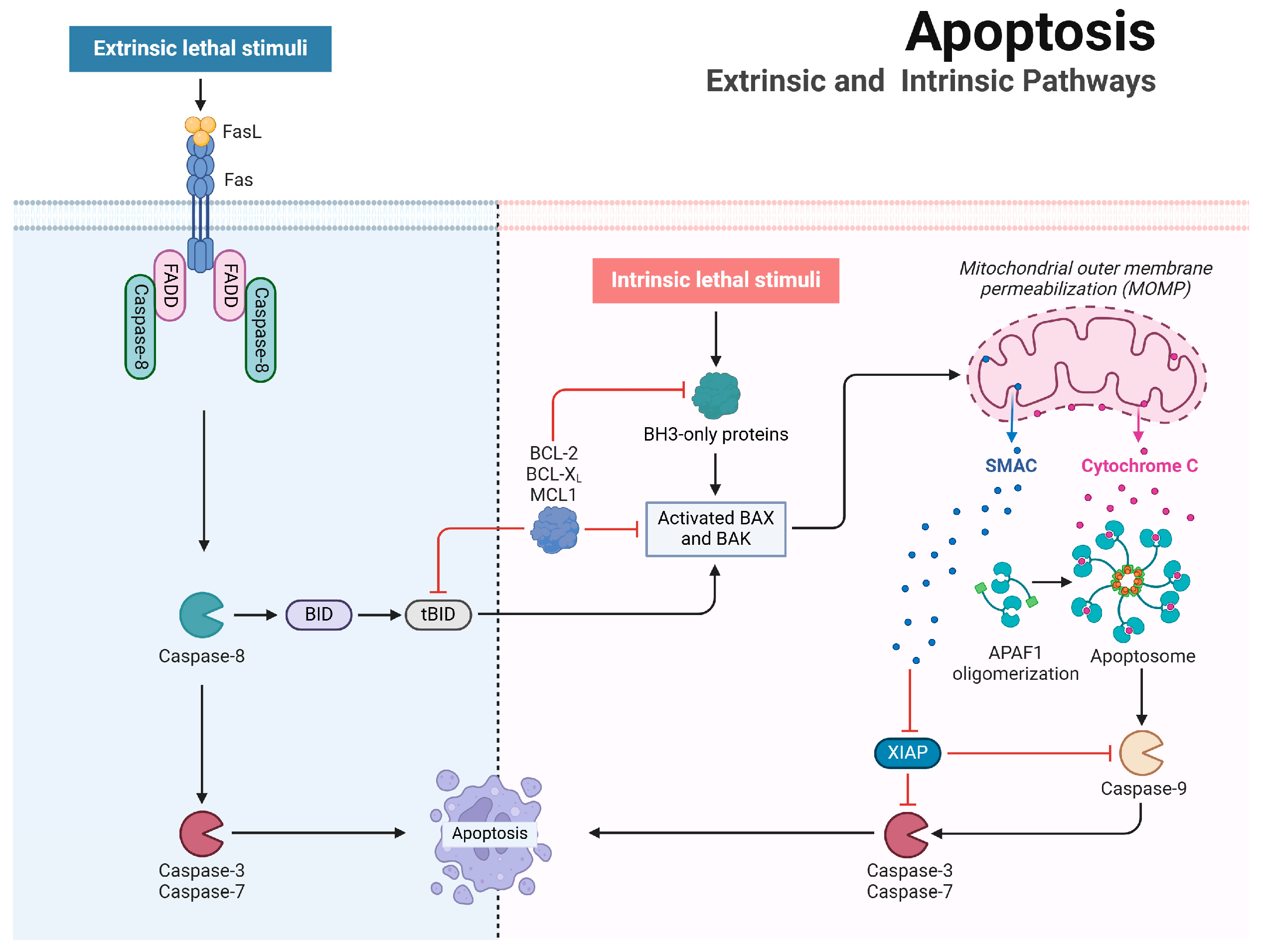
2. Apoptosis Mechanisms and Regulation
2.1. Mechanistic Insights into Apoptosis
2.2. Influenza A Virus Regulation of Apoptosis
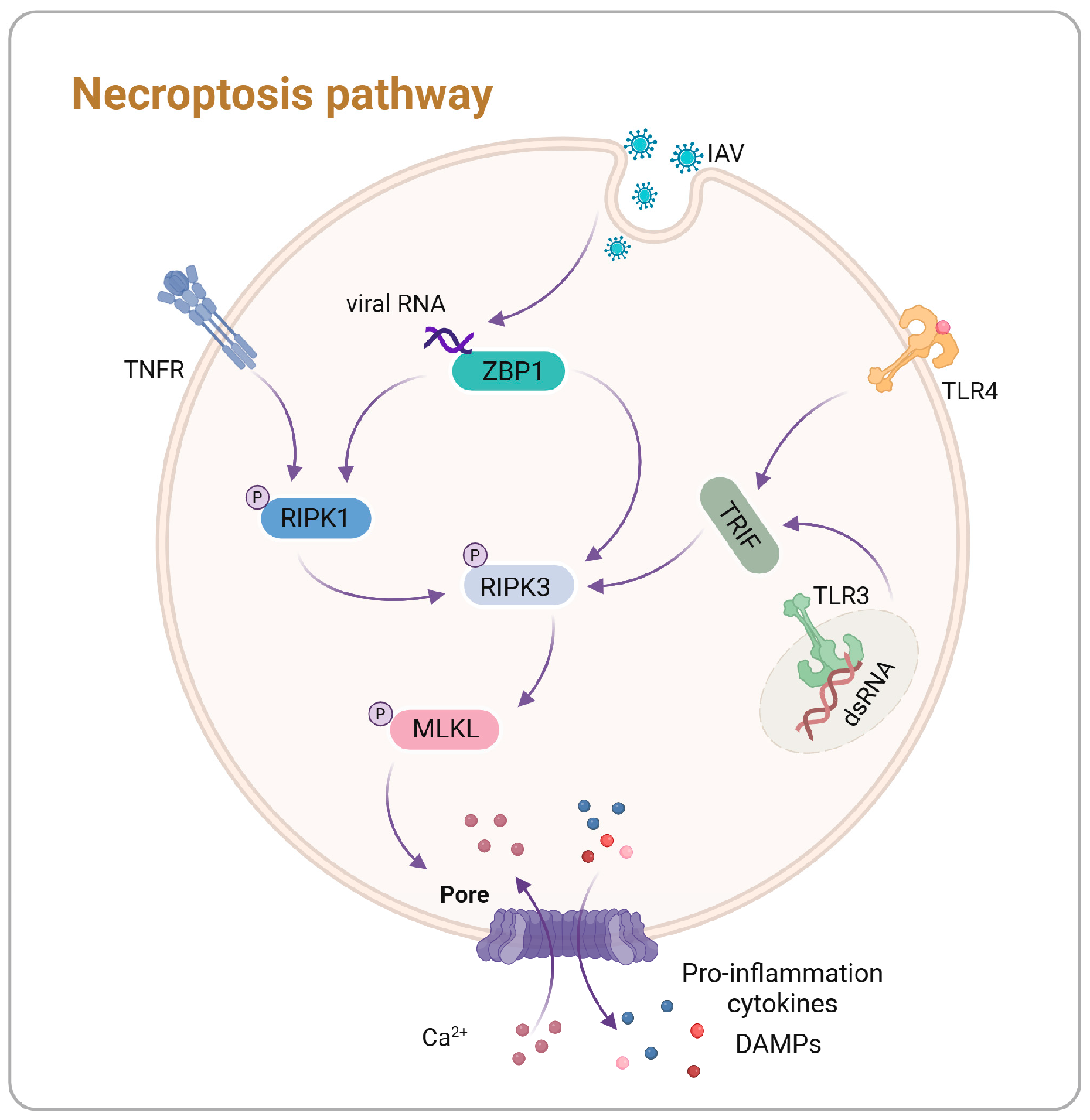
3. Necroptosis Mechanisms and Regulation
3.1. Mechanistic Insights into Necroptosis
3.2. Influenza A Virus Regulation of Necroptosis
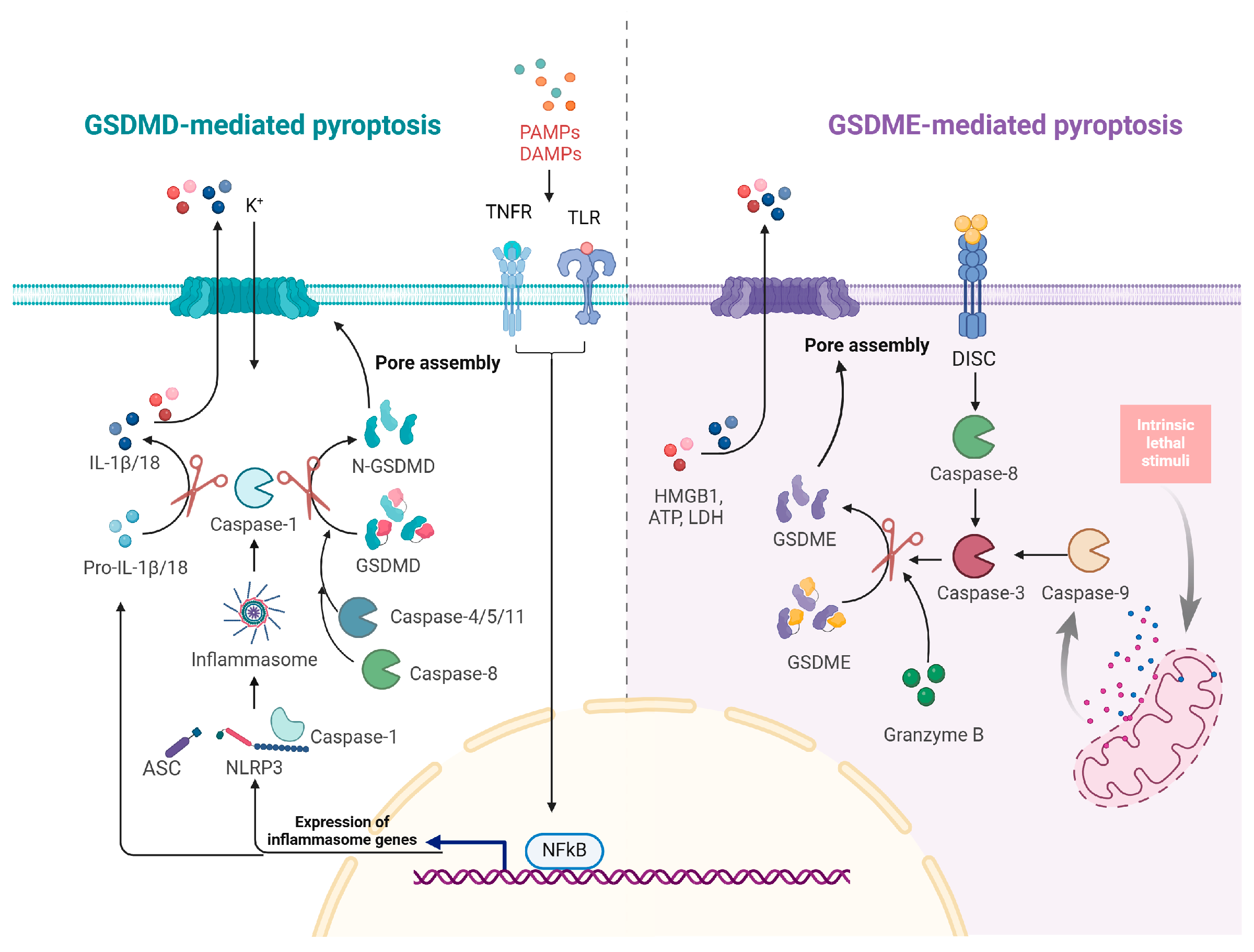
4. Pyroptosis Mechanism and Regulation
4.1. Mechanistic Insights into Pyroptosis
4.2. Influenza A Virus Regulation of Pyroptosis
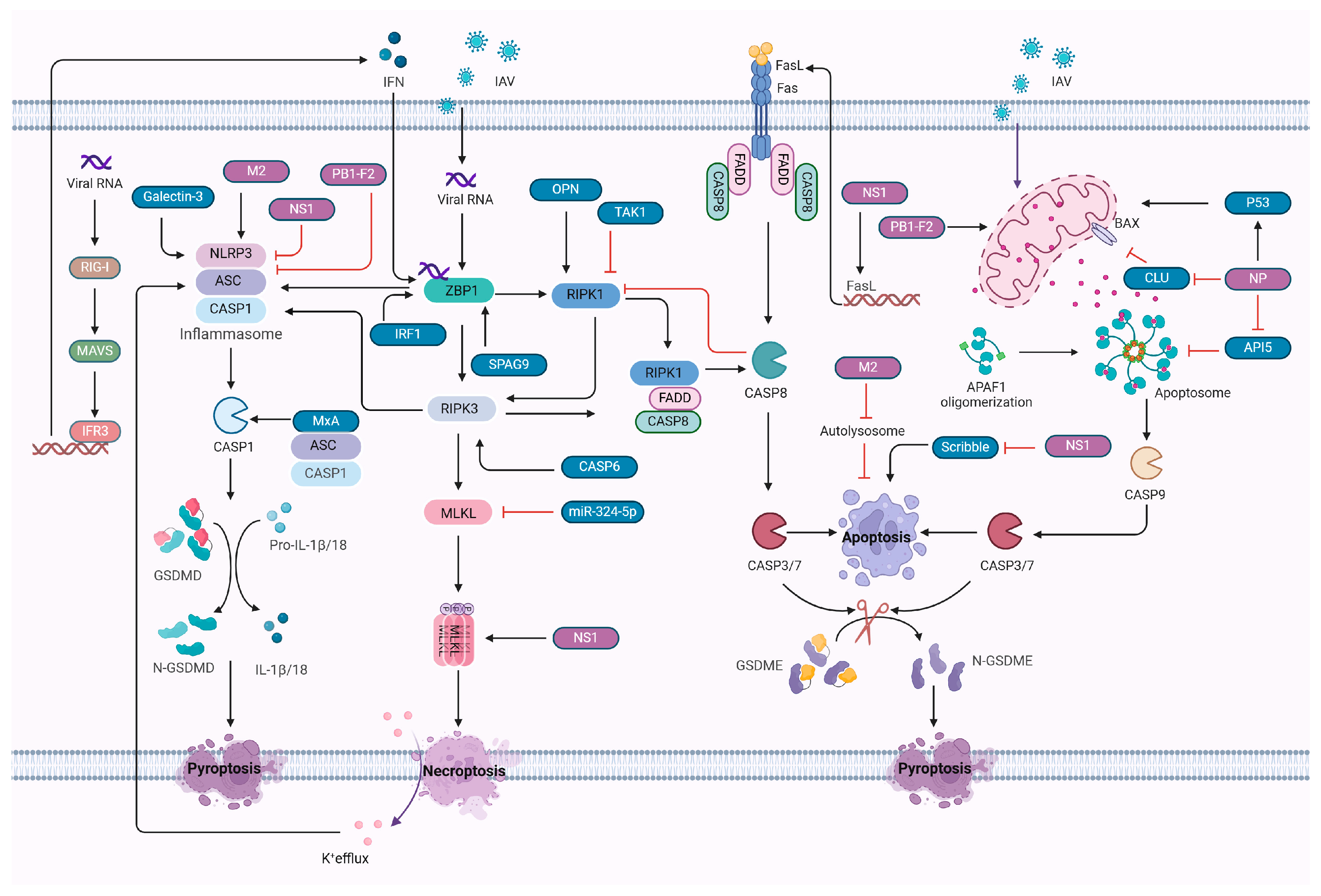
5. Activation and Interplay of Cell Death Pathways in Influenza A Virus Infection
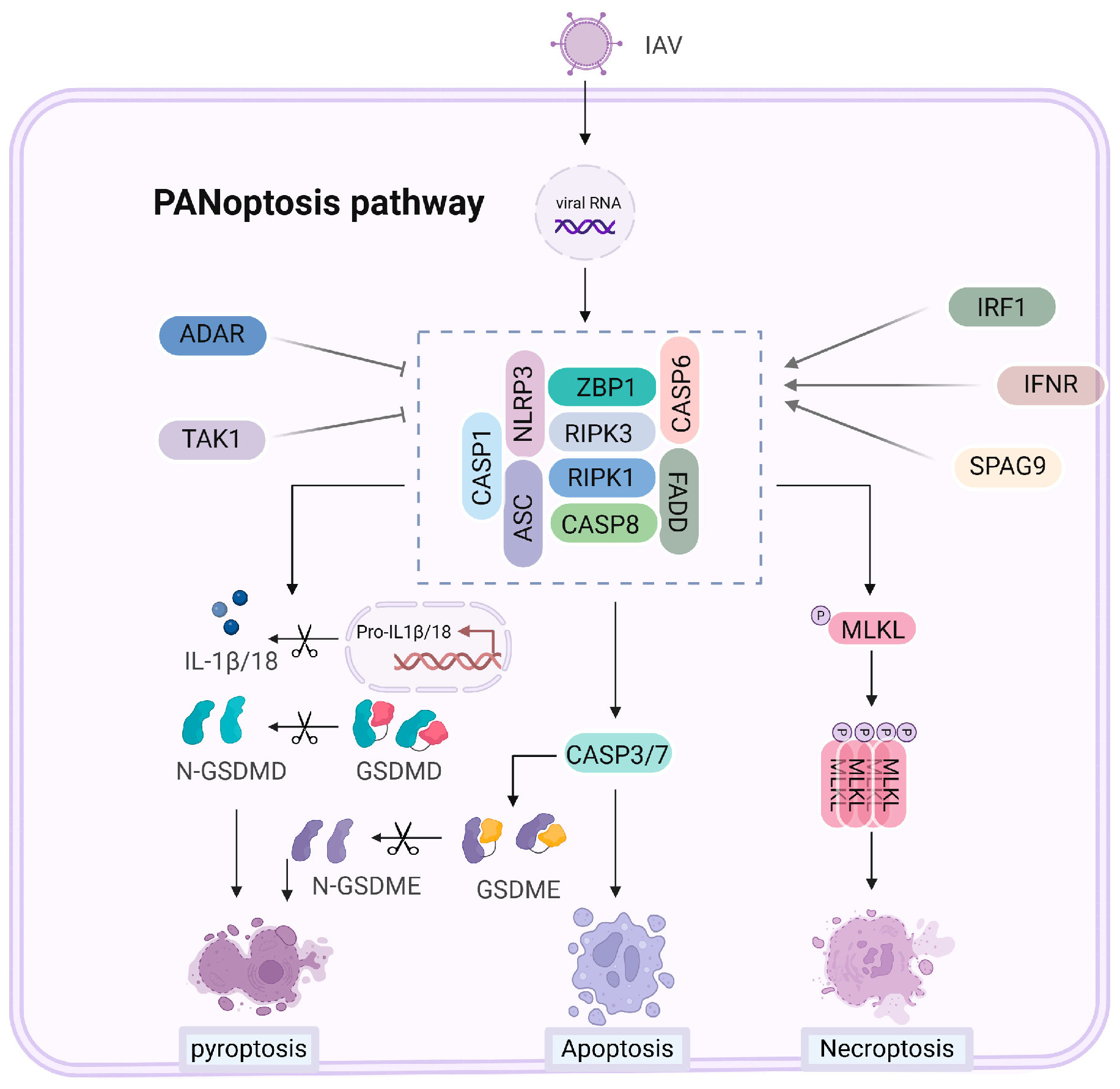
6. PANoptosis Mechanisms and Regulation with Influenza A Virus
6.1. Mechanisms of PANoptosis Activation
6.2. Influenza A Virus Regulation of PANoptosis
7. Cell Death and Inflammatory Response in IAV Infection
8. Therapeutic Strategies Targeting Cell Death in Influenza A Virus Infection
8.1. Small Molecule Inhibitors of Cell Death Pathways
8.1.1. Inhibition of NLRP3-Mediated Pyroptosis
8.1.2. Inhibition of RIPK1 and RIPK3-Mediated Necroptosis
8.1.3. Inhibition of Caspase-Mediated Apoptosis
8.2. Combination Therapies and Clinical Prospects
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of Global Seasonal Influenza-Associated Respiratory Mortality: A Modelling Study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Pandian, N.; Kanneganti, T.D. Panoptosis: A Unique Innate Immune Inflammatory Cell Death Modality. J. Immunol. 2022, 209, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for Datp and Cytochrome C. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. Bcl-2, Bcl-X(L) Sequester Bh3 Domain-Only Molecules Preventing Bax- and Bak-Mediated Mitochondrial Apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. Stepwise Activation of Bax and Bak by Tbid, Bim, and Puma Initiates Mitochondrial Apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a Heterodimeric Partner for Bcl-Xl and Bcl-2, Displaces Bax and Promotes Cell Death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-Dimensional Structure of the Apoptosome: Implications for Assembly, Procaspase-9 Binding, and Activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Cain, K.; Brown, D.G.; Langlais, C.; Cohen, G.M. Caspase Activation Involves the Formation of the Aposome, a Large (Approximately 700 Kda) Caspase-Activating Complex. J. Biol. Chem. 1999, 274, 22686–22692. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome C and Datp-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Ahmad, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Autoactivation of Procaspase-9 by Apaf-1-Mediated Oligomerization. Mol. Cell 1998, 1, 949–957. [Google Scholar] [CrossRef]
- Chandler, J.M.; Cohen, G.M.; MacFarlane, M. Different Subcellular Distribution of Caspase-3 and Caspase-7 Following Fas-Induced Apoptosis in Mouse Liver. J. Biol. Chem. 1998, 273, 10815–10818. [Google Scholar] [CrossRef] [PubMed]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human Inhibitor of Apoptosis Proteins: Why Xiap Is the Black Sheep of the Family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The Polypeptide Encoded by the Cdna for Human Cell Surface Antigen Fas Can Mediate Apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand Trail. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Bodmer, J.L.; Holler, N.; Mattmann, C.; Scuderi, P.; Terskikh, A.; Peitsch, M.C.; Tschopp, J. Characterization of Fas (Apo-1, Cd95)-Fas Ligand Interaction. J. Biol. Chem. 1997, 272, 18827–18833. [Google Scholar] [CrossRef]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. Trail Receptors 1 (Dr4) and 2 (Dr5) Signal Fadd-Dependent Apoptosis and Activate Nf-Kappab. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Strasser, A.; Jost, P.J.; Nagata, S. The Many Roles of Fas Receptor Signaling in the Immune System. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of Tnf Receptor I-Mediated Apoptosis Via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Nagata, S.; Tanaka, M. Programmed Cell Death and the Immune System. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A Novel Influenza a Virus Mitochondrial Protein that Induces Cell Death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza Virus Pb1-F2 Protein Induces Cell Death through Mitochondrial Ant3 and Vdac1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix Protein 2 of Influenza a Virus Blocks Autophagosome Fusion with Lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Nailwal, H.; Sharma, S.; Mayank, A.K.; Lal, S.K. The Nucleoprotein of Influenza a Virus Induces P53 Signaling and Apoptosis Via Attenuation of Host Ubiquitin Ligase Rnf43. Cell Death Dis. 2015, 6, e1768. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of Influenza a Virus Negatively Impacts Antiapoptotic Protein Api5 to Enhance E2f1-Dependent Apoptosis and Virus Replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza a Virus Nucleoprotein Induces Apoptosis in Human Airway Epithelial Cells: Implications of a Novel Interaction between Nucleoprotein and Host Protein Clusterin. Cell Death Dis. 2013, 4, e562. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza a Virus Ns1 Protein Prevents Activation of Nf-Kappab and Induction of Alpha/Beta Interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The Esev Pdz-Binding Motif of the Avian Influenza a Virus Ns1 Protein Protects Infected Cells from Apoptosis by Directly Targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef]
- Lam, W.Y.; Tang, J.W.; Yeung, A.C.; Chiu, L.C.; Sung, J.J.; Chan, P.K. Avian Influenza Virus a/Hk/483/97(H5n1) Ns1 Protein Induces Apoptosis in Human Airway Epithelial Cells. J. Virol. 2008, 82, 2741–2751. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. Dai (Dlm-1/Zbp1) Is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-Rnas Induce Zbp1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. Ripk3 Activates Parallel Pathways of Mlkl-Driven Necroptosis and Fadd-Mediated Apoptosis to Protect against Influenza a Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. Dai Senses Influenza a Virus Genomic Rna and Activates Ripk3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ji, L.; Liu, W.; Sun, J.; Liu, P.; Wang, X.; Liu, X.; Xu, X. Influenza Virus Infection Activates Tak1 to Suppress Ripk3-Independent Apoptosis and Ripk1-Dependent Necroptosis. Cell Commun. Signal 2024, 22, 372. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. Rip3 Mediates the Embryonic Lethality of Caspase-8-Deficient Mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic Activity of the Caspase-8-Flip(L) Complex Inhibits Ripk3-Dependent Necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Pierdomenico, M.; Negroni, A.; Stronati, L.; Vitali, R.; Prete, E.; Bertin, J.; Gough, P.J.; Aloi, M.; Cucchiara, S. Necroptosis Is Active in Children with Inflammatory Bowel Disease and Contributes to Heighten Intestinal Inflammation. Am. J. Gastroenterol. 2014, 109, 279–287. [Google Scholar] [CrossRef]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A Crucial Pathogenic Mediator of Human Disease. JCI Insight 2019, 4, 2. [Google Scholar] [CrossRef]
- Seo, J.; Nam, Y.W.; Kim, S.; Oh, D.B.; Song, J. Necroptosis Molecular Mechanisms: Recent Findings Regarding Novel Necroptosis Regulators. Exp. Mol. Med. 2021, 53, 1007–1017. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-Driven Assembly of the Rip1-Rip3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to Tnf-Alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in Tnf-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef]
- He, S.; Wang, X. Rip Kinases as Modulators of Inflammation and Immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. Rip3, an Energy Metabolism Regulator that Switches Tnf-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Dai/Zbp1/Dlm-1 Complexes with Rip3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus Vira. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Balachandran, S.; Mocarski, E.S. Viral Z-Rna Triggers Zbp1-Dependent Cell Death. Curr. Opin. Virol. 2021, 51, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Rall, G.F. Benefits and Perils of Necroptosis in Influenza Virus Infection. J. Virol. 2020, 94, 2. [Google Scholar] [CrossRef]
- Basavaraju, S.; Mishra, S.; Jindal, R.; Kesavardhana, S. Emerging Role of Zbp1 in Z-Rna Sensing, Influenza Virus-Induced Cell Death, and Pulmonary Inflammation. mBio 2022, 13, e0040122. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-Like Receptors Activate Programmed Necrosis in Macrophages through a Receptor-Interacting Kinase-3-Mediated Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-Like Receptor 3-Mediated Necrosis Via Trif, Rip3, and Mlkl. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. Ciaps Block Ripoptosome Formation, a Rip1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by Cflip Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, S.; Berghe, T.V.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The Induction and Consequences of Influenza a Virus-Induced Cell Death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Hulpiau, P.; Saeys, Y.; Bertrand, M.J.M.; Vandenabeele, P. An Evolutionary Perspective on the Necroptotic Pathway. Trends Cell Biol. 2016, 26, 721–732. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Gaba, A.; Xu, F.; Lu, Y.; Park, H.S.; Liu, G.; Zhou, Y. The Ns1 Protein of Influenza a Virus Participates in Necroptosis by Interacting with Mlkl and Increasing Its Oligomerization and Membrane Translocation. J. Virol. 2019, 93, 2. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Wang, Y.; Li, Y.; Shi, F.; Diao, H. Osteopontin Aggravates Acute Lung Injury in Influenza Virus Infection by Promoting Macrophages Necroptosis. Cell Death Discov. 2022, 8, 97. [Google Scholar] [CrossRef]
- Dou, X.; Yu, X.; Du, S.; Han, Y.; Li, L.; Zhang, H.; Yao, Y.; Du, Y.; Wang, X.; Li, J.; et al. Interferon-Mediated Repression of Mir-324-5p Potentiates Necroptosis to Facilitate Antiviral Defense. EMBO Rep. 2022, 23, e54438. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of Gsdmd by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kuang, S.; Zheng, J.; Yang, H.; Li, S.; Duan, S.; Shen, Y.; Ji, C.; Gan, J.; Xu, X.W.; Li, J. Structure Insight of Gsdmd Reveals the Basis of Gsdmd Autoinhibition in Cell Pyroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 10642–10647. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. Gsdmd Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Herr, D.R.; Yam, T.Y.A.; Tan, W.S.D.; Koh, S.S.; Wong, W.S.F.; Ong, W.Y.; Chayaburakul, K. Ultrastructural Characteristics of Dha-Induced Pyroptosis. Neuromolecular Med. 2020, 22, 293–303. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense against Infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular Lps. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of Tak1 Triggers Caspase-8-Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from Mlkl Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. Momp, Cell Suicide as a Bcl-2 Family Business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of Dfna5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. Zbp1/Dai Is an Innate Sensor of Influenza Virus Triggering the Nlrp3 Inflammasome and Programmed Cell Death Pathways. Sci. Immunol. 2016, 1, 2. [Google Scholar] [CrossRef]
- Tang, Z.; Mao, Y.; Ruan, P.; Li, J.; Qiu, X.; Meng, Y.; Wang, M.; Wu, G.; Wang, L.; Tan, Y. Drugs Targeting Cmpk2 Inhibit Pyroptosis to Alleviate Severe Pneumonia Caused by Multiple Respiratory Viruses. J. Med. Virol. 2024, 96, e29643. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Kuriakose, T.; Guy, C.S.; Samir, P.; Malireddi, R.K.S.; Mishra, A.; Kanneganti, T.D. Zbp1/Dai Ubiquitination and Sensing of Influenza Vrnps Activate Programmed Cell Death. J. Exp. Med. 2017, 214, 2217–2229. [Google Scholar] [CrossRef]
- Mishra, S.; Raj, A.S.; Kumar, A.; Rajeevan, A.; Kumari, P.; Kumar, H. Innate Immune Sensing of Influenza a Viral Rna through Ifi16 Promotes Pyroptotic Cell Death. iScience 2022, 25, 103714. [Google Scholar] [CrossRef]
- Lei, X.; Chen, Y.; Lien, E.; Fitzgerald, K.A. Mlkl-Driven Inflammasome Activation and Caspase-8 Mediate Inflammatory Cell Death in Influenza a Virus Infection. mBio 2023, 14, e0011023. [Google Scholar] [CrossRef]
- Ji, J.; Hou, J.; Xia, Y.; Xiang, Z.; Han, X. Nlrp3 Inflammasome Activation in Alveolar Epithelial Cells Promotes Myofibroblast Differentiation of Lung-Resident Mesenchymal Stem Cells during Pulmonary Fibrogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166077. [Google Scholar] [CrossRef]
- Kim, S.R.; Kim, D.I.; Kim, S.H.; Lee, H.; Lee, K.S.; Cho, S.H.; Lee, Y.C. Nlrp3 Inflammasome Activation by Mitochondrial Ros in Bronchial Epithelial Cells Is Required for Allergic Inflammation. Cell Death Dis. 2014, 5, e1498. [Google Scholar] [CrossRef]
- Lee, S.; Ishitsuka, A.; Noguchi, M.; Hirohama, M.; Fujiyasu, Y.; Petric, P.P.; Schwemmle, M.; Staeheli, P.; Nagata, K.; Kawaguchi, A. Influenza Restriction Factor Mxa Functions as Inflammasome Sensor in the Respiratory Epithelium. Sci. Immunol. 2019, 4, 2. [Google Scholar] [CrossRef]
- Chen, Y.J.; Wang, S.F.; Weng, I.C.; Hong, M.H.; Lo, T.H.; Jan, J.T.; Hsu, L.C.; Chen, H.Y.; Liu, F.T. Galectin-3 Enhances Avian H5n1 Influenza a Virus-Induced Pulmonary Inflammation by Promoting Nlrp3 Inflammasome Activation. Am. J. Pathol. 2018, 188, 1031–1042. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza Virus Activates Inflammasomes Via Its Intracellular M2 Ion Channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Cheung, P.H.; Ye, Z.W.; Lee, T.T.; Chen, H.; Chan, C.P.; Jin, D.Y. Pb1-F2 Protein of Highly Pathogenic Influenza a (H7n9) Virus Selectively Suppresses Rna-Induced Nlrp3 Inflammasome Activation through Inhibition of Mavs-Nlrp3 Interaction. J. Leukoc. Biol. 2020, 108, 1655–1663. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. Nek7 Is an Essential Mediator of Nlrp3 Activation Downstream of Potassium Efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Boal-Carvalho, I.; Mazel-Sanchez, B.; Silva, F.; Garnier, L.; Yildiz, S.; Bonifacio, J.P.; Niu, C.; Williams, N.; Francois, P.; Schwerk, N.; et al. Influenza a Viruses Limit Nlrp3-Nek7-Complex Formation and Pyroptosis in Human Macrophages. EMBO Rep. 2020, 21, e50421. [Google Scholar] [CrossRef]
- Moriyama, M.; Chen, I.Y.; Kawaguchi, A.; Koshiba, T.; Nagata, K.; Takeyama, H.; Hasegawa, H.; Ichinohe, T. The Rna- and Trim25-Binding Domains of Influenza Virus Ns1 Protein Are Essential for Suppression of Nlrp3 Inflammasome-Mediated Interleukin-1beta Secretion. J. Virol. 2016, 90, 4105–4114. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Wan, X.; Li, J.; Wang, Y.; Yu, X.; He, X.; Shi, J.; Deng, G.; Zeng, X.; Tian, G.; Li, Y.; et al. H7n9 Virus Infection Triggers Lethal Cytokine Storm by Activating Gasdermin E-Mediated Pyroptosis of Lung Alveolar Epithelial Cells. Natl. Sci. Rev. 2022, 9, nwab137. [Google Scholar] [CrossRef]
- Guy, C.; Baran, M.; Ribo-Molina, P.; van den Hoogen, B.G.; Bowie, A.G. Viral Sensing by Epithelial Cells Involves Pkr- and Caspase-3-Dependent Generation of Gasdermin E Pores. iScience 2023, 26, 107698. [Google Scholar] [CrossRef]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. Ninj1 Mediates Plasma Membrane Rupture during Lytic Cell Death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- David, L.; Borges, J.P.; Hollingsworth, L.R.; Volchuk, A.; Jansen, I.; Garlick, E.; Steinberg, B.E.; Wu, H. Ninj1 Mediates Plasma Membrane Rupture by Cutting and Releasing Membrane Disks. Cell 2024, 187, 2224–2235.e16. [Google Scholar] [CrossRef] [PubMed]
- Malik, G.; Zhou, Y. Innate Immune Sensing of Influenza a Virus. Viruses 2020, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.D. Irf1 Is a Transcriptional Regulator of Zbp1 Promoting Nlrp3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 2018, 200, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Gui, R.; Zheng, H.; Ma, L.; Liu, R.; Lin, X.; Ke, X.; Ye, C.; Jian, X.; Chen, Q. Sperm-Associated Antigen 9 Promotes Influenza a Virus-Induced Cell Death Via the C-Jun N-Terminal Kinase Signaling Pathway. mBio 2022, 13, e0061522. [Google Scholar] [CrossRef] [PubMed]
- Tao, P.; Sun, J.; Wu, Z.; Wang, S.; Wang, J.; Li, W.; Pan, H.; Bai, R.; Zhang, J.; Wang, Y.; et al. A Dominant Autoinflammatory Disease Caused by Non-Cleavable Variants of Ripk1. Nature 2020, 577, 109–114. [Google Scholar] [CrossRef]
- Gullett, J.M.; Tweedell, R.E.; Kanneganti, T.D. It’s All in the Pan: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by Panoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells 2022, 11, 2. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Burton, A.R.; Porter, S.N.; Vogel, P.; Pruett-Miller, S.M.; Kanneganti, T.D. The Zalpha2 Domain of Zbp1 Is a Molecular Switch Regulating Influenza-Induced Panoptosis and Perinatal Lethality during Development. J. Biol. Chem. 2020, 295, 8325–8330. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Rajesh, Y.; Kanneganti, T.D. Immune Regulator Irf1 Contributes to Zbp1-, Aim2-, Ripk1-, and Nlrp12-Panoptosome Activation and Inflammatory Cell Death (Panoptosis). J. Biol. Chem. 2023, 299, 105141. [Google Scholar] [CrossRef]
- de Reuver, R.; Verdonck, S.; Dierick, E.; Nemegeer, J.; Hessmann, E.; Ahmad, S.; Jans, M.; Blancke, G.; Van Nieuwerburgh, F.; Botzki, A.; et al. Adar1 Prevents Autoinflammation by Suppressing Spontaneous Zbp1 Activation. Nature 2022, 607, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Muendlein, H.I.; Connolly, W.M.; Magri, Z.; Smirnova, I.; Ilyukha, V.; Gautam, A.; Degterev, A.; Poltorak, A. Zbp1 Promotes Lps-Induced Cell Death and Il-1beta Release Via Rhim-Mediated Interactions with Ripk1. Nat. Commun. 2021, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Deng, W.; Bai, Y.; Miao, R.; Mei, S.; Zhang, Z.; Pan, Y.; Wang, Y.; Min, R.; Deng, F.; et al. The Lysosomal Rag-Ragulator Complex Licenses Ripk1 and Caspase-8-Mediated Pyroptosis by Yersinia. Science 2021, 372, 2. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- Shinya, K.; Gao, Y.; Cilloniz, C.; Suzuki, Y.; Fujie, M.; Deng, G.; Zhu, Q.; Fan, S.; Makino, A.; Muramoto, Y.; et al. Integrated Clinical, Pathologic, Virologic, and Transcriptomic Analysis of H5n1 Influenza Virus-Induced Viral Pneumonia in the Rhesus Macaque. J. Virol. 2012, 86, 6055–6066. [Google Scholar] [CrossRef]
- Kim, K.S.; Jung, H.; Shin, I.K.; Choi, B.R.; Kim, D.H. Induction of Interleukin-1 Beta (Il-1beta) Is a Critical Component of Lung Inflammation during Influenza a (H1n1) Virus Infection. J. Med. Virol. 2015, 87, 1104–1112. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Sharma, B.R.; Bynigeri, R.R.; Wang, Y.; Lu, J.; Kanneganti, T.D. Zbp1 Drives Iav-Induced Nlrp3 Inflammasome Activation and Lytic Cell Death, Panoptosis, Independent of the Necroptosis Executioner Mlkl. Viruses 2023, 15, 11. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of Tnf-Alpha and Ifn-Gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, J.; Zhou, D.; Zhang, S.; Wang, L.; Tian, Q.; Li, C.; Liu, J.; Wu, Y.; Li, J.; et al. Trim34 Modulates Influenza Virus-Activated Programmed Cell Death by Targeting Z-DNA-Binding Protein 1 for K63-Linked Polyubiquitination. J. Biol. Chem. 2022, 298, 101611. [Google Scholar] [CrossRef]
- Karki, R.; Lee, S.; Mall, R.; Pandian, N.; Wang, Y.; Sharma, B.R.; Malireddi, R.S.; Yang, D.; Trifkovic, S.; Steele, J.A.; et al. Zbp1-Dependent Inflammatory Cell Death, Panoptosis, and Cytokine Storm Disrupt Ifn Therapeutic Efficacy during Coronavirus Infection. Sci. Immunol. 2022, 7, eabo6294. [Google Scholar] [CrossRef]
- Rodrigue-Gervais, I.G.; Labbe, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular Inhibitor of Apoptosis Protein Ciap2 Protects against Pulmonary Tissue Necrosis during Influenza Virus Infection to Promote Host Survival. Cell Host Microbe 2014, 15, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Boyd, D.F.; Nikhar, S.; Zhang, T.; Siokas, I.; Van de Velde, L.A.; Gaevert, J.; Meliopoulos, V.; Thapa, B.; Rodriguez, D.A.; et al. Necroptosis Blockade Prevents Lung Injury in Severe Influenza. Nature 2024, 628, 835–843. [Google Scholar] [CrossRef]
- Lin, Y.H.; Platt, M.P.; Gilley, R.P.; Brown, D.; Dube, P.H.; Yu, Y.; Gonzalez-Juarbe, N. Influenza Causes Mlkl-Driven Cardiac Proteome Remodeling during Convalescence. Circ. Res. 2021, 128, 570–584. [Google Scholar] [CrossRef]
- Qin, C.; Sai, X.Y.; Qian, X.F.; Wu, Y.; Zou, L.F.; Wang, H.M.; Bian, T.; Yan, Z. Close Relationship between Ciap2 and Human Ards Induced by Severe H7n9 Infection. Biomed. Res. Int. 2019, 2019, 2121357. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The Nlrp3 Inflammasome Mediates in Vivo Innate Immunity to Influenza a Virus through Recognition of Viral Rna. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Samir, P.; Zheng, M.; Malireddi, R.K.S.; Karki, R.; Sharma, B.R.; Place, D.E.; Briard, B.; Vogel, P.; Kanneganti, T.D. Ddx3x Coordinates Host Defense against Influenza Virus by Activating the Nlrp3 Inflammasome and Type I Interferon Response. J. Biol. Chem. 2021, 296, 100579. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The Intracellular Sensor Nlrp3 Mediates Key Innate and Healing Responses to Influenza a Virus Via the Regulation of Caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Wu, S.; Cai, J.; Yang, Y.; Ren, X.; Feng, Y.; Chen, L.; Qin, B.; Xu, C.; Yang, H.; et al. The H7n9 Influenza a Virus Infection Results in Lethal Inflammation in the Mammalian Host Via the Nlrp3-Caspase-1 Inflammasome. Sci. Rep. 2017, 7, 7625. [Google Scholar] [CrossRef]
- Corry, J.; Kettenburg, G.; Upadhyay, A.A.; Wallace, M.; Marti, M.M.; Wonderlich, E.R.; Bissel, S.J.; Goss, K.; Sturgeon, T.J.; Watkins, S.C.; et al. Infiltration of Inflammatory Macrophages and Neutrophils and Widespread Pyroptosis in Lung Drive Influenza Lethality in Nonhuman Primates. PLoS Pathog. 2022, 18, e1010395. [Google Scholar] [CrossRef]
- Rosli, S.; Harpur, C.M.; Lam, M.; West, A.C.; Hodges, C.; Mansell, A.; Lawlor, K.E.; Tate, M.D. Gasdermin D Promotes Hyperinflammation and Immunopathology during Severe Influenza a Virus Infection. Cell Death Dis. 2023, 14, 727. [Google Scholar] [CrossRef]
- Chu, H.; Shuai, H.; Hou, Y.; Zhang, X.; Wen, L.; Huang, X.; Hu, B.; Yang, D.; Wang, Y.; Yoon, C.; et al. Targeting Highly Pathogenic Coronavirus-Induced Apoptosis Reduces Viral Pathogenesis and Disease Severity. Sci. Adv. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, W.; Zhang, J.; Du, Y.; Fang, M.; Zhang, A.; Sungcad, G.; Chon, S.; Xing, J. Loss of Trim29 Mitigates Viral Myocarditis by Attenuating Perk-Driven Er Stress Response in Male Mice. Nat. Commun. 2024, 15, 3481. [Google Scholar] [CrossRef]
- Oltean, T.; Van San, E.; Divert, T.; Berghe, T.V.; Saelens, X.; Maelfait, J.; Takahashi, N.; Vandenabeele, P. Viral Dosing of Influenza a Infection Reveals Involvement of Ripk3 and Fadd, but Not Mlkl. Cell Death Dis. 2021, 12, 471. [Google Scholar] [CrossRef]
- Oltean, T.; Maelfait, J.; Saelens, X.; Vandenabeele, P. Need for Standardization of Influenza a Virus-Induced Cell Death in Vivo to Improve Consistency of Inter-Laboratory Research Findings. Cell Death Discov. 2024, 10, 247. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. Mcc950 Directly Targets the Nlrp3 Atp-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. Pb1-F2 Peptide Derived from Avian Influenza a Virus H7n9 Induces Inflammation Via Activation of the Nlrp3 Inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Dai, M.Y.; Huang, Y.; Ren, X.C.; Jiang, M.L.; Qiao, J.P.; Zhang, W.Y.; Xu, Y.H.; Shen, J.L.; Zhang, R.Q.; et al. Influenza a Virus Triggers Acute Exacerbation of Chronic Obstructive Pulmonary Disease by Increasing Proinflammatory Cytokines Secretion Via Nlrp3 Inflammasome Activation. J. Inflamm. 2022, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N Protein Promotes Nlrp3 Inflammasome Activation to Induce Hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Amand, M.; Adams, P.; Schober, R.; Iserentant, G.; Servais, J.Y.; Moutschen, M.; Seguin-Devaux, C. The Anti-Caspase 1 Inhibitor Vx-765 Reduces Immune Activation, Cd4(+) T Cell Depletion, Viral Load, and Total Hiv-1 DNA in Hiv-1 Infected Humanized Mice. Elife 2023, 12, e83207. [Google Scholar] [CrossRef]
- He, Z.; An, S.; Chen, J.; Zhang, S.; Tan, C.; Yu, J.; Ye, H.; Wu, Y.; Yuan, J.; Wu, J.; et al. Neural Progenitor Cell Pyroptosis Contributes to Zika Virus-Induced Brain Atrophy and Represents a Therapeutic Target. Proc. Natl. Acad. Sci. USA 2020, 117, 23869–23878. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. Fda-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Adrover, J.M.; Carrau, L.; Dassler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; R tenOever, B.; et al. Disulfiram Inhibits Neutrophil Extracellular Trap Formation and Protects Rodents from Acute Lung Injury and SARS-CoV-2 Infection. JCI Insight 2022, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Bosio, G.; Malberti, F.; Cuzzoli, A.; Testa, S.; Romanini, L.; Fioravanti, A.; Morandini, A.; Pianta, L.; Giannotti, G.; et al. Canakinumab as Treatment for COVID-19-Related Pneumonia: A Prospective Case-Control Study. Int. J. Infect. Dis. 2021, 104, 433–440. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.W. Anakinra as a Potential Treatment for COVID-19. Drugs Today 2023, 59, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Zhang, L.; Richner, J.M.; Class, J.; Rehman, J.; Malik, A.B. Interleukin-1ra Mitigates SARS-CoV-2-Induced Inflammatory Lung Vascular Leakage and Mortality in Humanized K18-Hace-2 Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2773–2785. [Google Scholar] [CrossRef]
- Sichelstiel, A.; Yadava, K.; Trompette, A.; Salami, O.; Iwakura, Y.; Nicod, L.P.; Marsland, B.J. Targeting Il-1beta and Il-17a Driven Inflammation during Influenza-Induced Exacerbations of Chronic Lung Inflammation. PLoS ONE 2014, 9, e98440. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, B.; Hong, S.; Cai, Y. A Case Report and Literature Review on Tocilizumab-Cured Acute Necrotizing Encephalopathy Caused by Influenza a Virus. Front. Pediatr. 2024, 12, 1351478. [Google Scholar] [CrossRef]
- Koh, J.C.; Murugasu, A.; Krishnappa, J.; Thomas, T. Favorable Outcomes with Early Interleukin 6 Receptor Blockade in Severe Acute Necrotizing Encephalopathy of Childhood. Pediatr. Neurol. 2019, 98, 80–84. [Google Scholar] [CrossRef]
- Xu, G.; Li, Y.; Zhang, S.; Peng, H.; Wang, Y.; Li, D.; Jin, T.; He, Z.; Tong, Y.; Qi, C.; et al. SARS-CoV-2 Promotes Ripk1 Activation to Facilitate Viral Propagation. Cell Res. 2021, 31, 1230–1243. [Google Scholar] [CrossRef]
- Lin, B.; Jin, Z.; Chen, X.; Zhao, L.; Weng, C.; Chen, B.; Tang, Y.; Lin, L. Necrostatin-1 Protects Mice from Acute Lung Injury by Suppressing Necroptosis and Reactive Oxygen Species. Mol. Med. Rep. 2020, 21, 2171–2181. [Google Scholar] [CrossRef]
- Mago, E.; Zhao, X.; Zhang, W.; Shao, Q.; Li, P.; Huang, S.; Ding, X.; Liu, H.; Sun, T.; He, F.; et al. Rip1 Kinase Inactivation Protects against Lps-Induced Acute Respiratory Distress Syndrome in Mice. Int. Immunopharmacol. 2024, 133, 112060. [Google Scholar] [CrossRef]
- Zhou, Y.; Cai, Z.; Zhai, Y.; Yu, J.; He, Q.; He, Y.; Jitkaew, S.; Cai, Z. Necroptosis Inhibitors: Mechanisms of Action and Therapeutic Potential. Apoptosis 2024, 29, 22–44. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.N.; To, K.K.W.; Chan, J.F.W.; Cheng, V.C.C.; Liu, K.S.H.; Tam, A.; Chan, T.C.; Zhang, A.J.; Li, P.; Wong, T.L.; et al. Efficacy of Clarithromycin-Naproxen-Oseltamivir Combination in the Treatment of Patients Hospitalized for Influenza a(H3n2) Infection: An Open-Label Randomized, Controlled, Phase Iib/Iii Trial. Chest 2017, 151, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Liu, B.; Bao, L.; Lv, Q.; Li, F.; Li, H.; An, Y.; Zhang, X.; Cao, B.; Wang, C. Delayed Oseltamivir Plus Sirolimus Treatment Attenuates H1n1 Virus-Induced Severe Lung Injury Correlated with Repressed Nlrp3 Inflammasome Activation and Inflammatory Cell Infiltration. PLoS Pathog. 2018, 14, e1007428. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables Nlrp3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. Rip3 Induces Apoptosis Independent of Pronecrotic Kinase Activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of Keratinocyte Necroptosis Mediated by Ripk1/Ripk3/Mlkl Provides a Protective Effect against Psoriatic Inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef]
- He, F.; Zheng, G.; Hu, J.; Ge, W.; Ji, X.; Bradley, J.L.; Peberdy, M.A.; Ornato, J.P.; Tang, W. Necrosulfonamide Improves Post-Resuscitation Myocardial Dysfunction Via Inhibiting Pyroptosis and Necroptosis in a Rat Model of Cardiac Arrest. Eur. J. Pharmacol. 2022, 926, 175037. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; Jiang, N.; He, D.; Bai, Y.; Xin, Y. Rapamycin Combined with Mcc950 to Treat Multiple Sclerosis in Experimental Autoimmune Encephalomyelitis. J. Cell Biochem. 2019, 120, 5160–5168. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Apoptosis | Necroptosis | Pyroptosis |
|---|---|---|---|
| DNA Fragmentation | Yes | No | Yes |
| Chromatin Condensation | Yes (pyknotic nuclei) | No | Minimal or absent |
| Membrane Integrity | Yes (intact until late stage) | No | No |
| Organelle Swelling | No | Yes | Yes |
| Caspase Activation | Yes (Caspase-3, -7, -9, etc.) | No (RIPK1/RIPK3/MLKL pathway) | Yes (Caspase-1, -4, -5, -11, -8, -3) |
| Cell Death Mechanism | Programmed | Programmed | Programmed |
| Inflammatory Nature | No (immunologically silent) | Yes (highly inflammatory, DAMP release) | Yes (highly inflammatory, IL-1β, IL-18 release) |
| Viral Protein | Mechanism of Action | Effect on Apoptosis | References |
|---|---|---|---|
| PB1-F2 | Localizes to mitochondria, induces mitochondrial membrane permeabilization | Activates intrinsic apoptotic pathway | [20,21] |
| M2 | Interacts with ATG5/Beclin-1 complex | Inhibits autophagosome fusion, promotes apoptosis indirectly | [22] |
| NP | Stabilizes p53, downregulates API5 | Amplifies apoptosis, reduces anti-apoptotic signals | [23,24] |
| NP | Reduces CLU–Bax interaction | Facilitates mitochondrial apoptosis | [25] |
| NS1 | Inhibits type I IFN response, binds to Scribble | Suppresses apoptosis, enhances viral persistence | [26,27] |
| NS1 (H5N1) | Upregulates FasL mRNA expression | Promotes apoptosis | [28] |
| Drug Name | Target Protein/ Pathway | Efficacy | References |
|---|---|---|---|
| MCC950 | NLRP3 | Reduces inflammation and lung injury in models of IAV and SARS-CoV-2. | [125,126,127,128] |
| Z25, Z08 | CMPK2 | Reduces pulmonary inflammation in RSV and potentially IAV. | [76] |
| VX-765 | Caspase-1 | Inhibits pyroptosis and reduces inflammation during viral infections. | [129,130] |
| Disulfiram | GSDMD | Mitigates acute lung injury and improves survival in TRALI and SARS-CoV-2 models. | [131,132] |
| Canakinumab | IL-1β | Controls severe inflammation in pneumonia associated with SARS-CoV-2. | [133] |
| Anakinra | IL-1 Receptor | Reduces severe inflammatory responses in SARS-CoV-2 and IAV infection. | [134,135,136] |
| Tocilizumab | IL-6 Receptor | Improves clinical outcomes in acute necrotizing encephalopathy from H1N1 virus. | [137,138] |
| Necrostatin-1, Nec-1s | RIPK1 | Protects against lung injury by inhibiting RIPK1-RIPK3 signaling. | [139,140,141] |
| GSK’872, GSK’843, GSK’840 | RIPK3 | Effective in mitigating necroptosis in preclinical models. | [142] |
| Necrosulfonamide | MLKL | Exhibits protective effects in necroptosis-related pathologies. | [112] |
| z-LEHD-fmk | Caspase-9 | Improves survival and reduces lung injury in respiratory virus models. | [121] |
| Clarithromycin + Naproxen + Oseltamivir | Antiviral + anti-inflammatory | Reduces viral titers and mortality in H3N2-infected patients. | [143] |
| Oseltamivir + Sirolimus | mTOR-NF-κB-NLRP3 | Reduces pro-inflammatory cytokines and alleviates lung injury in H1N1 pdm09 infection. | [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Liu, K. Mechanistic Insights into Influenza A Virus-Induced Cell Death and Emerging Treatment Strategies. Vet. Sci. 2024, 11, 555. https://doi.org/10.3390/vetsci11110555
Sun Y, Liu K. Mechanistic Insights into Influenza A Virus-Induced Cell Death and Emerging Treatment Strategies. Veterinary Sciences. 2024; 11(11):555. https://doi.org/10.3390/vetsci11110555
Chicago/Turabian StyleSun, Yuling, and Kaituo Liu. 2024. "Mechanistic Insights into Influenza A Virus-Induced Cell Death and Emerging Treatment Strategies" Veterinary Sciences 11, no. 11: 555. https://doi.org/10.3390/vetsci11110555
APA StyleSun, Y., & Liu, K. (2024). Mechanistic Insights into Influenza A Virus-Induced Cell Death and Emerging Treatment Strategies. Veterinary Sciences, 11(11), 555. https://doi.org/10.3390/vetsci11110555