Transcriptional Profiles of Secondary Metabolite Biosynthesis Genes and Cytochromes in the Leaves of Four Papaver Species


 ,
,  , ,
, ,
Abstract
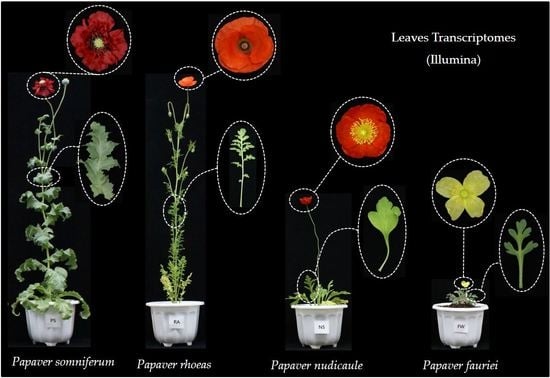
1. Introduction
2. Results
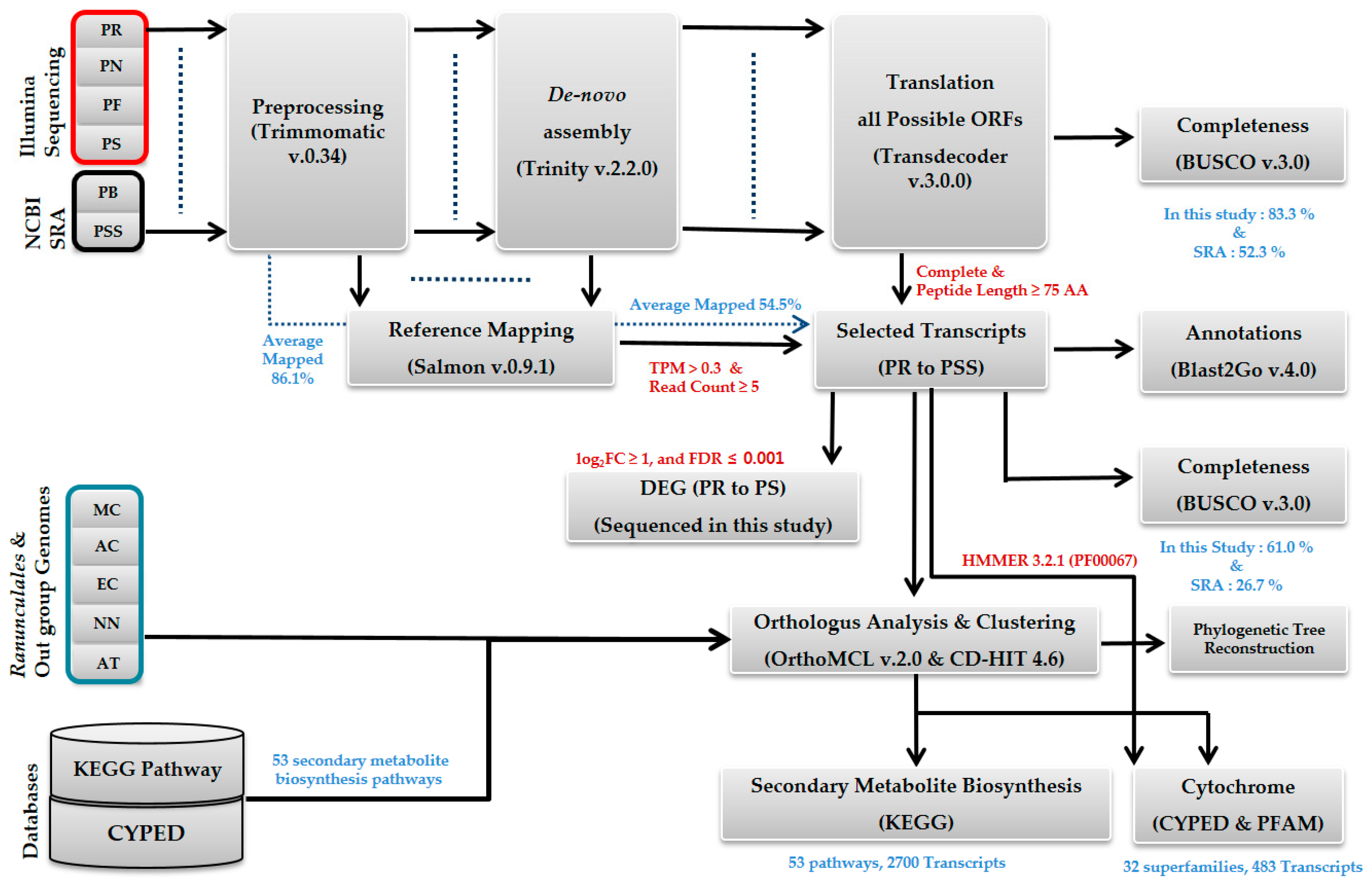
2.1. Sequencing and De Novo Transcriptome Assembly
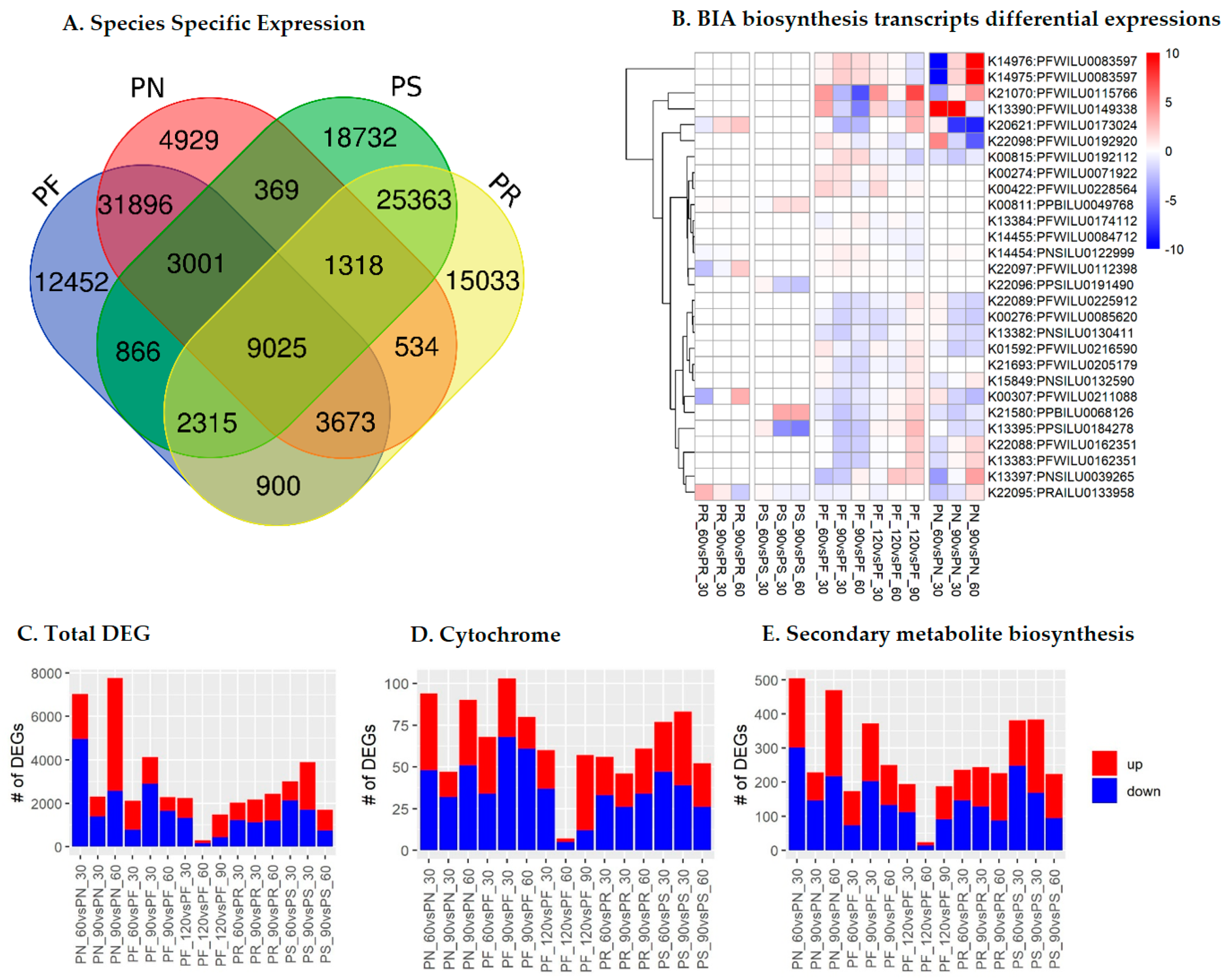
2.2. Species- and Stage-Specific Differentially Expressed Transcripts
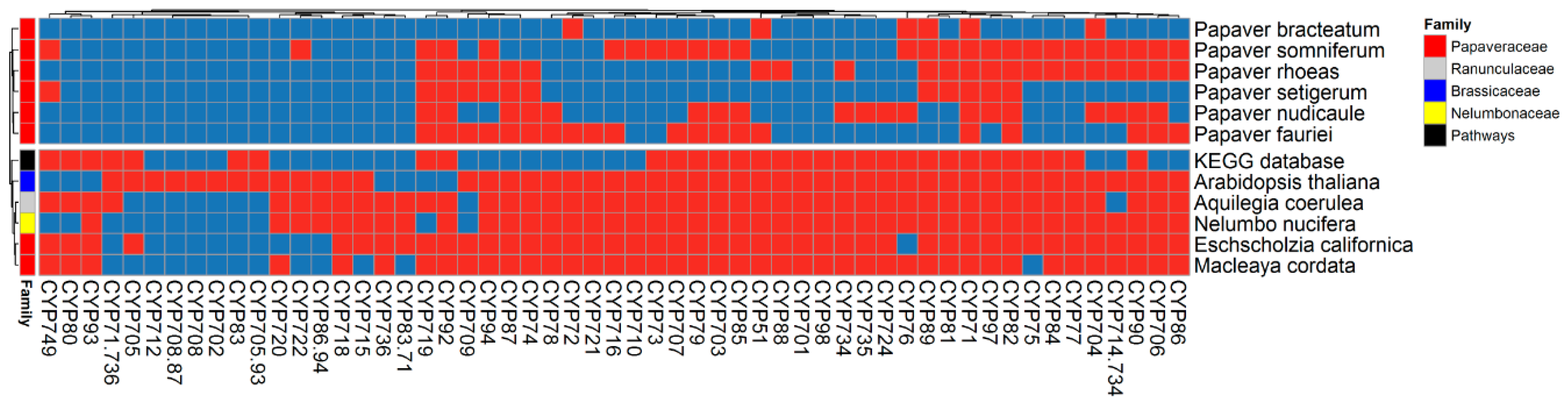
2.3. Cytochrome Profiles
2.4. Cytochromes Involved in Secondary Metabolite Biosynthesis (SMB)
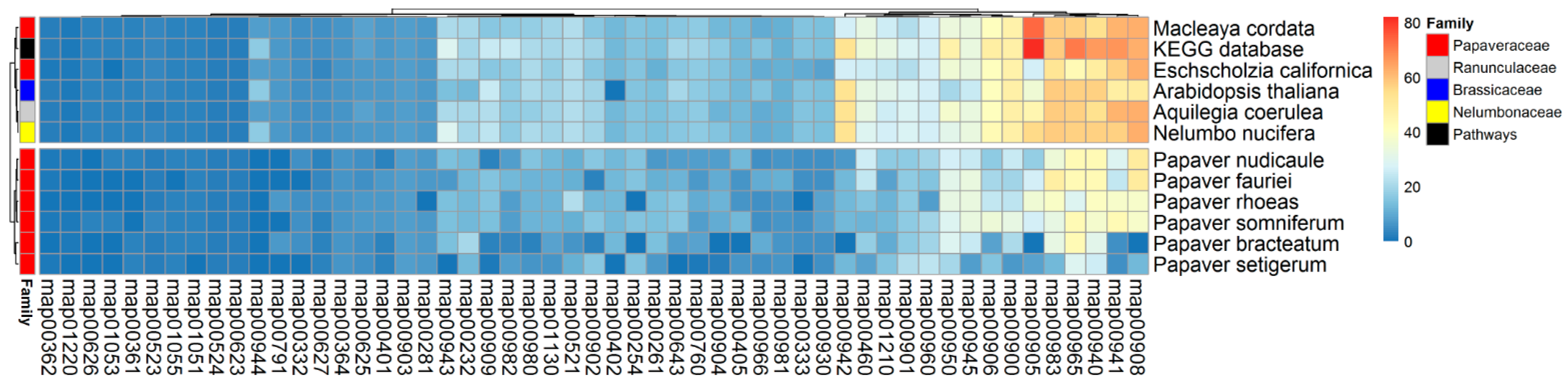
2.5. SMB Pathway Profiles
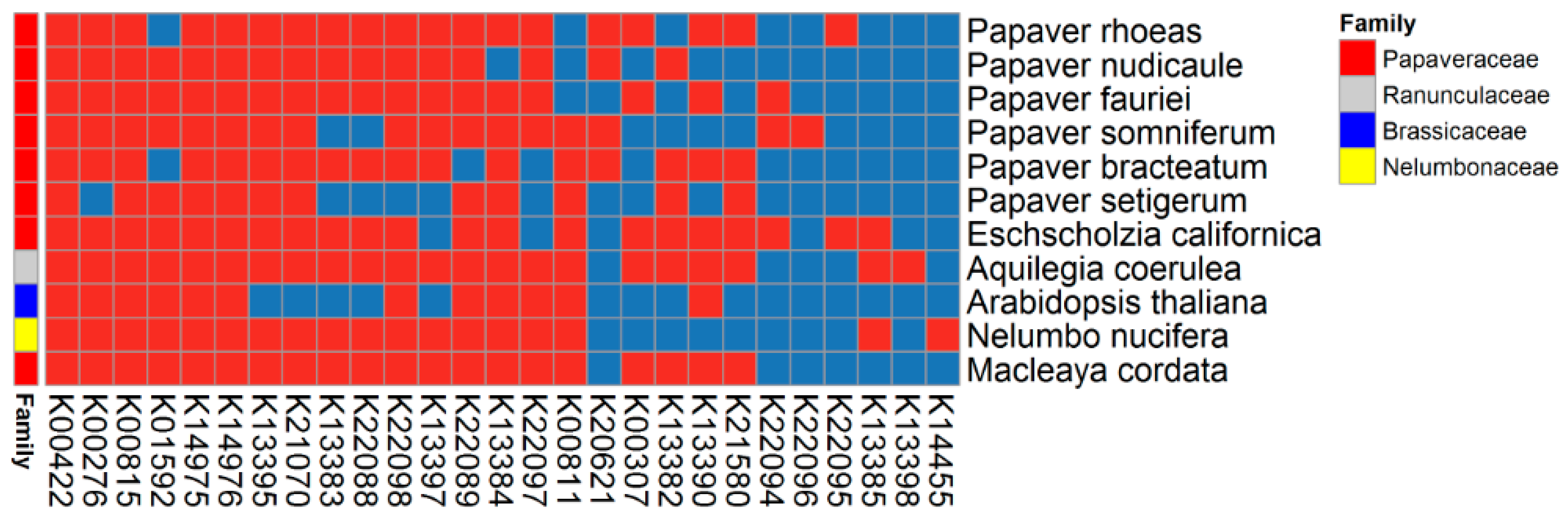
2.6. BIA Biosynthesis Pathway
3. Discussion
4. Material and Methods
4.1. Plant Samples
4.2. Illumina Library Preparation and Sequencing
4.3. Assembly and Annotation
4.4. LC-QTOF-MS/MS Metabolomic Analysis
4.5. Differential Gene Expression
4.6. KEGG SMB Proteins
4.7. Ortholog Analysis and Phylogeny Construction
4.8. Cytochrome Family Analysis
4.9. Data Submission
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trang, T.; Al-Hasani, R.; Salvemini, D.; Salter, M.W.; Gutstein, H.; Cahill, C.M. Pain and Poppies: The Good, the Bad, and the Ugly of Opioid Analgesics. J. Neurosci. 2015, 35, 13879–13888. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi-Kia, F.; Lorigooini, Z.; Amini-Khoei, H. Medicinal plants: Past history and future perspective. J. Herbmed. Pharmacol. 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Mathiyalagan, R.; Yang, D.C. Ginseng nanoparticles: A budding tool for cancer treatment. Nanomedicine 2017, 12, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B.; et al. Update of the Scientific Opinion on opium alkaloids in poppy seeds. EFSA J. 2018, 16, e05243. [Google Scholar]
- Barceloux, D.G. Heroin and the Opium Poppy Plant (Papaver somniferum L.). In Medical Toxicology of Drug Abuse; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 546–578. [Google Scholar] [CrossRef]
- Bernath, J. Poppy: The Genus Papaver; Harwood Academic Publishers: London, UK, 1999; Volume 3, p. 390. [Google Scholar]
- Winzer, T.; Kern, M.; King, A.J.; Larson, T.R.; Teodor, R.I.; Donninger, S.L.; Li, Y.; Dowle, A.A.; Cartwright, J.; Bates, R.; et al. Morphinan biosynthesis in opium poppy requires a P450-oxidoreductase fusion protein. Science 2015, 349, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Filsinger Interrante, M.; Smolke, C.D. Complete biosynthesis of opioids in yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Smolke, C.D. Engineering biosynthesis of the anticancer alkaloid noscapine in yeast. Nat. Commun. 2016, 7, 12137. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Winzer, T.; Yang, X.; Li, Y.; Ning, Z.; He, Z.; Teodor, R.; Lu, Y.; Bowser, T.A.; Graham, I.A.; et al. The opium poppy genome and morphinan production. Science 2018. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Leitch, I.J.; Anadon-Rosell, A.; Canela, M.Á.; Gálvez, F.; Garnatje, T.; Gras, A.; Hidalgo, O.; Johnston, E.; Mas de Xaxars, G.; et al. Recent updates and developments to plant genome size databases. Nucleic Acids Res. 2014, 42, D1159–D1166. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Lavania, U.C. Evolutionary DNA variation in Papaver. Genome 1991, 34, 763–768. [Google Scholar] [CrossRef]
- Boke, H.; Ozhuner, E.; Turktas, M.; Parmaksiz, I.; Ozcan, S.; Unver, T. Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnol. J. 2015, 13, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Desgagné-Penix, I.; Farrow, S.C.; Cram, D.; Nowak, J.; Facchini, P.J. Integration of deep transcript and targeted metabolite profiles for eight cultivars of opium poppy. Plant Mol. Boil. 2012, 79, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Desgagné-Penix, I.; Khan, M.F.; Schriemer, D.C.; Cram, D.; Nowak, J.; Facchini, P.J. Integration of deep transcriptome and proteome analyses reveals the components of alkaloid metabolism in opium poppy cell cultures. BMC Plant Boil. 2010, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Lakhwani, D.; Gupta, P.; Mishra, B.K.; Shukla, S.; Asif, M.H.; Trivedi, P.K. Comparative Transcriptome Analysis Using High Papaverine Mutant of Papaver somniferum Reveals Pathway and Uncharacterized Steps of Papaverine Biosynthesis. PLoS ONE 2013, 8, e65622. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Shin, Y.; Ha, I.; Lee, M.; Lee, S.-G.; Kang, B.-C.; Kyeong, D.; Kim, D. Transcriptome Profiling of Two Ornamental and Medicinal Papaver Herbs. Int. J. Mol. Sci. 2018, 19, 3192. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R. Cytochrome P450 diversity in the tree of life. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Werck-Reichhart, D. A P450-centric view of plant evolution. Plant J. 2011, 66, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. KEGG Bioinformatics Resource for Plant Genomics and Metabolomics. In Plant Bioinformatics: Methods and Protocols; Edwards, D., Ed.; Springer: New York, NY, USA, 2016; pp. 55–70. [Google Scholar] [CrossRef]
- Dastmalchi, M.; Park, M.R.; Morris, J.S.; Facchini, P. Family portraits: The enzymes behind benzylisoquinoline alkaloid diversity. Phytochem. Rev. 2018, 17, 249–277. [Google Scholar] [CrossRef]
- Durst, F.; Nelson David, R. Diversity and Evolution of Plant P450 and P450-Reductases. Drug Metab. Drug Interact. 1995, 12, 189–206. [Google Scholar] [CrossRef]
- Liao, W.; Zhao, S.; Zhang, M.; Dong, K.; Chen, Y.; Fu, C.; Yu, L. Transcriptome Assembly and Systematic Identification of Novel Cytochrome P450s in Taxus chinensis. Front. Plant Sci. 2017, 8, 1468. [Google Scholar] [CrossRef] [PubMed]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859. [Google Scholar] [CrossRef] [PubMed]
- Devi, B.S.R.; Kim, Y.-J.; Sathiyamoorthy, S.; Khorolragchaa, A.; Gayathri, S.; Parvin, S.; Yang, D.-U.; Selvi, S.K.; Lee, O.R.; Lee, S.; et al. Classification and characterization of putative cytochrome P450 genes from Panax ginseng C. A. Meyer. Biochemistry 2011, 76, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyam, S.; Mathiyalagan, R.; Natarajan, S.; Kim, Y.-J.; Jang, M.-G.; Park, J.-H.; Yang, D.C. Transcript expression profiling for adventitious roots of Panax ginseng Meyer. Gene 2014, 546, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Morris, J.S.; Lee, E.-J.; Desgagné-Penix, I.; Bross, C.D.; Chang, L.; Chen, X.; Farrow, S.C.; Zhang, Y.; Soh, J.; et al. Transcriptome analysis of 20 taxonomically related benzylisoquinoline alkaloid-producing plants. BMC Plant Boil. 2015, 15, 227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, Y.; Huang, P.; Ma, Y.; Qing, Z.; Tang, Q.; Cao, H.; Cheng, P.; Zheng, Y.; Yuan, Z.; et al. The Genome of Medicinal Plant Macleaya cordata Provides New Insights into Benzylisoquinoline Alkaloids Metabolism. Mol. Plant 2017, 10, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Yamada, Y.; Purwanto, R.; Minakuchi, Y.; Toyoda, A.; Hirakawa, H.; Sato, F. Mining of the Uncharacterized Cytochrome P450 Genes Involved in Alkaloid Biosynthesis in California Poppy Using a Draft Genome Sequence. Plant Cell Physiol. 2018, 59, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Sharma, I.; Pati, P.K. Versatile roles of brassinosteroid in plants in the context of its homoeostasis, signaling and crosstalks. Front. Plant Sci. 2015, 6, 950. [Google Scholar] [CrossRef] [PubMed]
- Moses, T.; Pollier, J.; Thevelein, J.M.; Goossens, A. Bioengineering of plant (tri)terpenoids: From metabolic engineering of plants to synthetic biology in vivo and in vitro. New Phytol. 2013, 200, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Gutensohn, M.; Klempien, A.; Kaminaga, Y.; Nagegowda, D.A.; Negre-Zakharov, F.; Huh, J.-H.; Luo, H.; Weizbauer, R.; Mengiste, T.; Tholl, D.; et al. Role of aromatic aldehyde synthase in wounding/herbivory response and flower scent production in different Arabidopsis ecotypes. Plant J. 2011, 66, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Morant, M.; Jørgensen, K.; Schaller, H.; Pinot, F.; Møller, B.L.; Werck-Reichhart, D.; Bak, S. CYP703 Is an Ancient Cytochrome P450 in Land Plants Catalyzing in-Chain Hydroxylation of Lauric Acid to Provide Building Blocks for Sporopollenin Synthesis in Pollen. Plant Cell 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Irmisch, S.; Clavijo McCormick, A.; Günther, J.; Schmidt, A.; Boeckler, G.A.; Gershenzon, J.; Unsicker, S.B.; Köllner, T.G. Herbivore-induced poplar cytochrome P450 enzymes of the CYP71 family convert aldoximes to nitriles which repel a generalist caterpillar. Plant J. 2014, 80, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Pinot, F.; Beisson, F. Cytochrome P450 metabolizing fatty acids in plants: Characterization and physiological roles. FEBS J. 2010, 278, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, C.; Li-Beisson, Y.; Philippar, K. Fatty Acid and Lipid Transport in Plant Cells. Trends Plant Sci. 2016, 21, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Mei, W.; Qin, Y.; Song, W.; Li, J.; Zhu, Y. Cotton GhPOX1 encoding plant class III peroxidase may be responsible for the high level of reactive oxygen species production that is related to cotton fiber elongation. J. Genet. Genom. 2009, 36, 141–150. [Google Scholar] [CrossRef]
- Zhong, R.; Morrison, W.H.; Himmelsbach, D.S.; Poole, F.L.; Ye, Z.-H. Essential Role of Caffeoyl Coenzyme A O-Methyltransferase in Lignin Biosynthesis in Woody Poplar Plants. Plant Physiol. 2000, 124, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Choi, H.; An, G. Roles of lignin biosynthesis and regulatory genes in plant development. J. Integr. Plant Boil. 2015, 57, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, S. Biosynthesis and Regulation of Phenylpropanoids in Plants. Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq: Reference generation and analysis with Trinity. Nat. Protoc. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Boil. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—Reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Boil. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Boil. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Boil. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Gricman, Ł.; Vogel, C.; Pleiss, J. Identification of universal selectivity-determining positions in cytochrome P450 monooxygenases by systematic sequence-based literature mining. Proteins Struct. Funct. Bioinform. 2015, 83, 1593–1603. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Papaver rhoeas (PR) | Papaver nudicaule (PN) | Papaver fauriei (PF) | Papaver somniferum (PS) |
|---|---|---|---|---|
| Sequenced bases (Gb) | 69.92 (Gb) | 61.32 (Gb) | 78.76 (Gb) | 54.79 (Gb) |
| Assembled transcripts (Mb) | 434,296 (267 Mb) | 203,097 (157 Mb) | 280,706 (206 Mb) | 219,124 (183 Mb) |
| Selected transcripts (Mb) | 27,973 (49 Mb) | 26,371 (50 Mb) | 34,294 (60 Mb) | 32,598 (65 Mb) |
| Blast hits (%) | 22,041 (78.8%) | 21,664 (82.2%) | 27,263 (79.5%) | 26,872 (82.4%) |
| Map | Pathway | CYP Superfamilies |
|---|---|---|
| map00905 | Brassinosteroid biosynthesis | CYP716, CYP749, CYP92, CYP90, CYP724 |
| map00904 | Diterpenoid biosynthesis | CYP701, CYP708.87, CYP82 |
| map00950 | Isoquinoline alkaloid biosynthesis | CYP719, CYP80, CYP82 |
| map00906 | Carotenoid biosynthesis | CYP97, CYP707 |
| map00940 | Phenylpropanoid biosynthesis | CYP84, CYP73 |
| map00460 | Cyanoamino acid metabolism | CYP79 |
| map00966 | Glucosinolate biosynthesis | CYP79 |
| map00941 | Flavonoid biosynthesis | CYP75 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Jung, M.; Ha, I.J.; Lee, M.Y.; Lee, S.-G.; Shin, Y.; Subramaniyam, S.; Oh, J. Transcriptional Profiles of Secondary Metabolite Biosynthesis Genes and Cytochromes in the Leaves of Four Papaver Species. Data 2018, 3, 55. https://doi.org/10.3390/data3040055
Kim D, Jung M, Ha IJ, Lee MY, Lee S-G, Shin Y, Subramaniyam S, Oh J. Transcriptional Profiles of Secondary Metabolite Biosynthesis Genes and Cytochromes in the Leaves of Four Papaver Species. Data. 2018; 3(4):55. https://doi.org/10.3390/data3040055
Chicago/Turabian StyleKim, Dowan, Myunghee Jung, In Jin Ha, Min Young Lee, Seok-Geun Lee, Younhee Shin, Sathiyamoorthy Subramaniyam, and Jaehyeon Oh. 2018. "Transcriptional Profiles of Secondary Metabolite Biosynthesis Genes and Cytochromes in the Leaves of Four Papaver Species" Data 3, no. 4: 55. https://doi.org/10.3390/data3040055
APA StyleKim, D., Jung, M., Ha, I. J., Lee, M. Y., Lee, S.-G., Shin, Y., Subramaniyam, S., & Oh, J. (2018). Transcriptional Profiles of Secondary Metabolite Biosynthesis Genes and Cytochromes in the Leaves of Four Papaver Species. Data, 3(4), 55. https://doi.org/10.3390/data3040055