De Novo Transcriptome Assembly of Cucurbita Pepo L. Leaf Tissue Infested by Aphis Gossypii

 , ,
, , 
Abstract
:1. Summary
2. Data Description
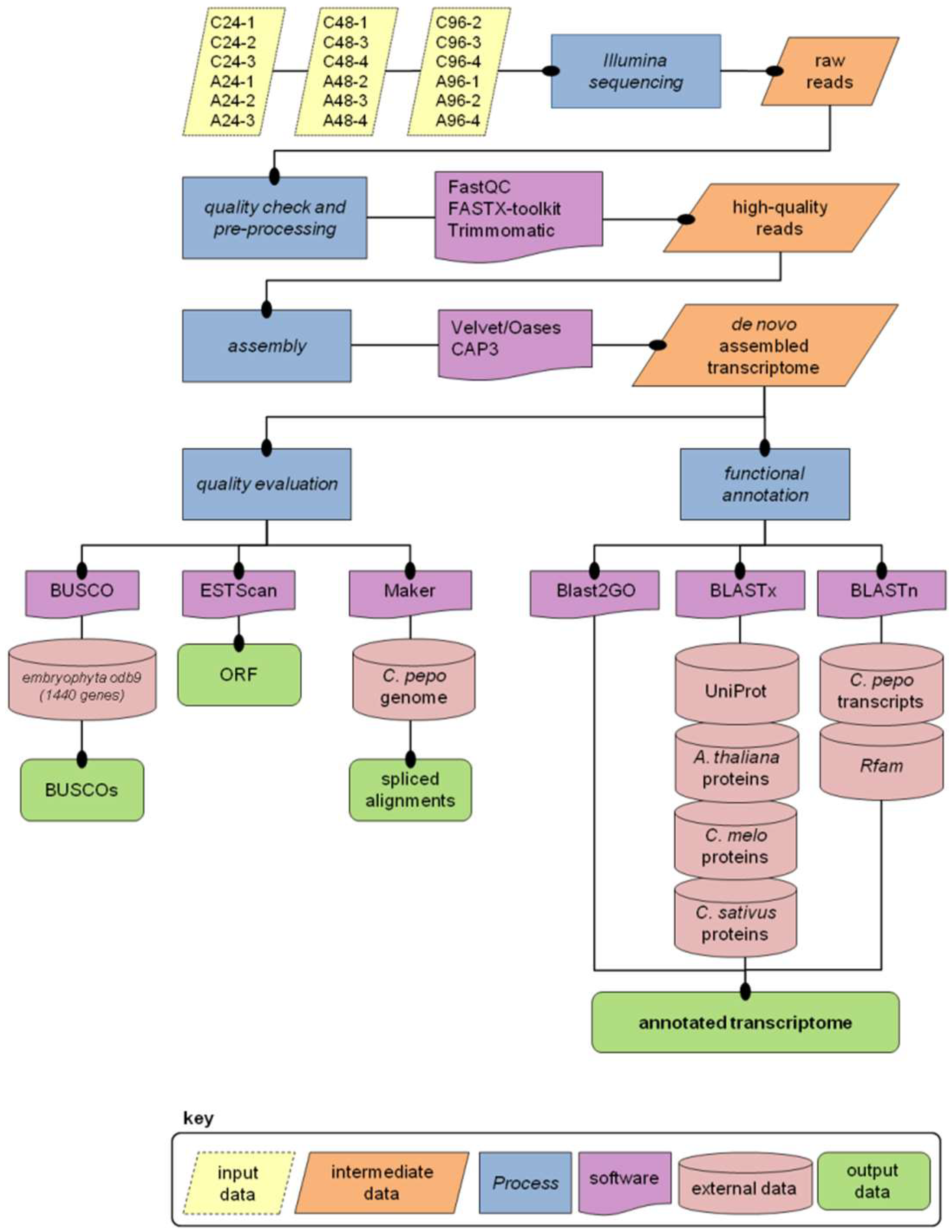
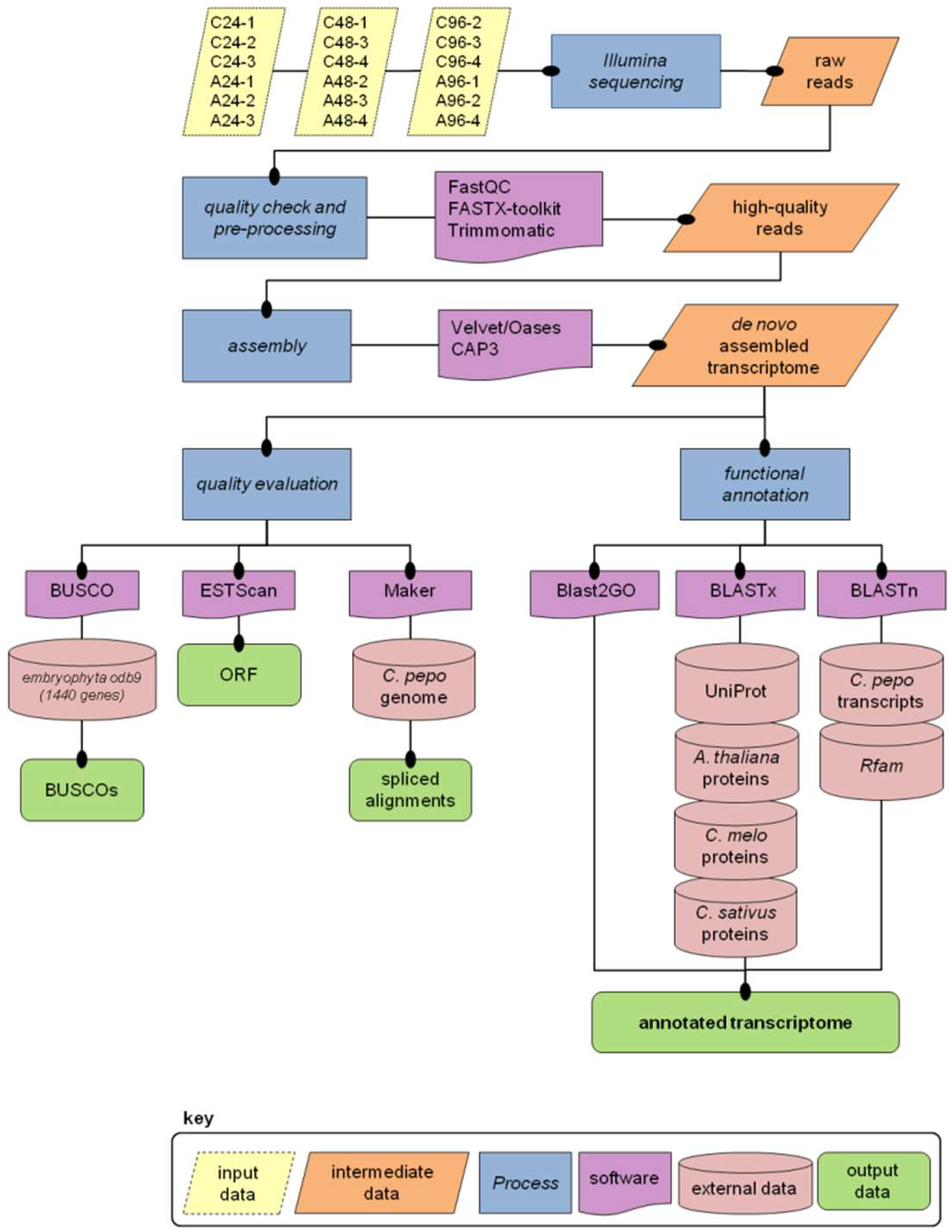
2.1. Illumina Read Processing and Transcriptome Assembly
2.2. Annotation
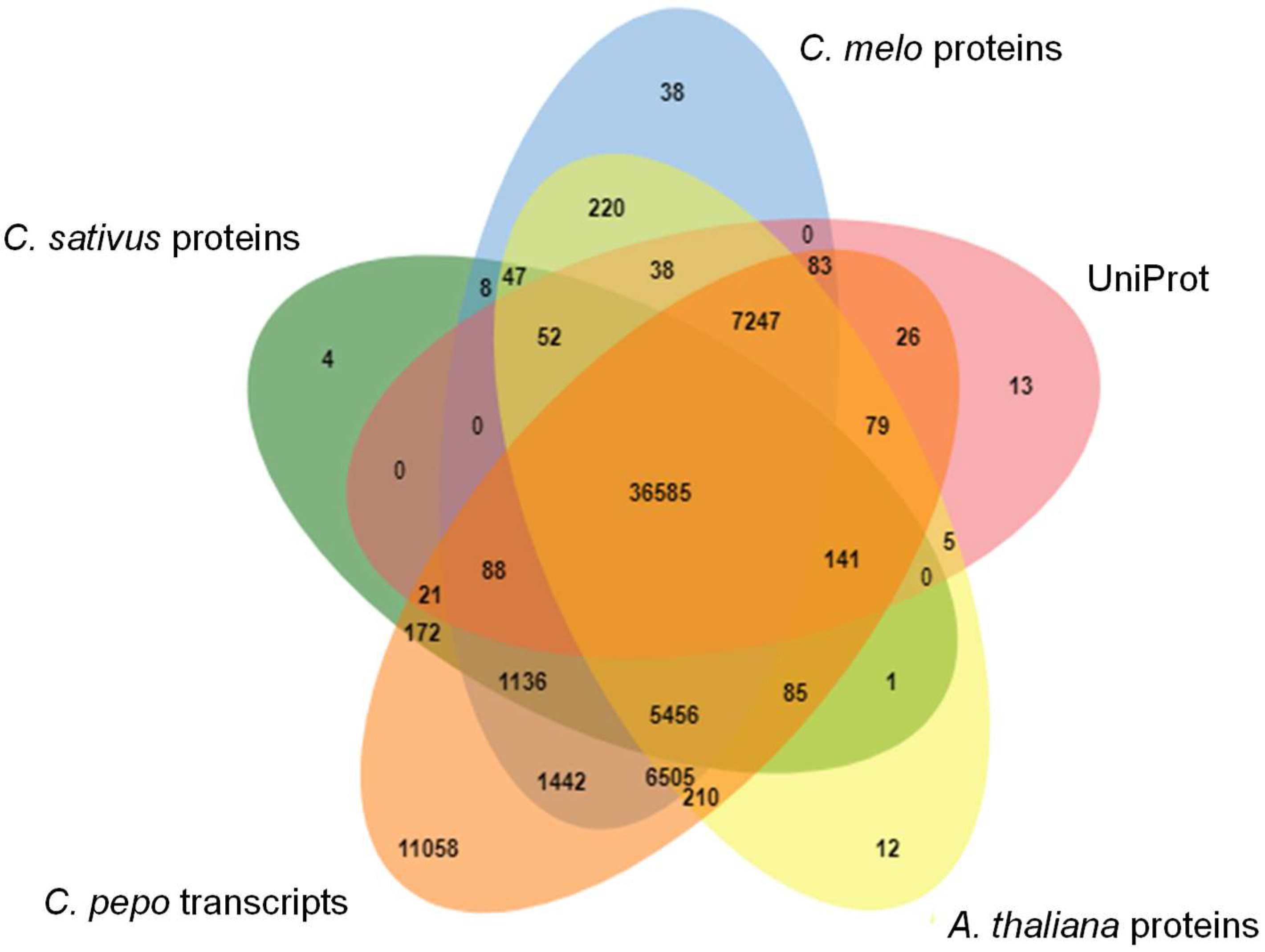
2.3. Evaluation of Transcriptome Quality and Completeness
2.4. Value of the Data
2.5. Data Records
3. Methods
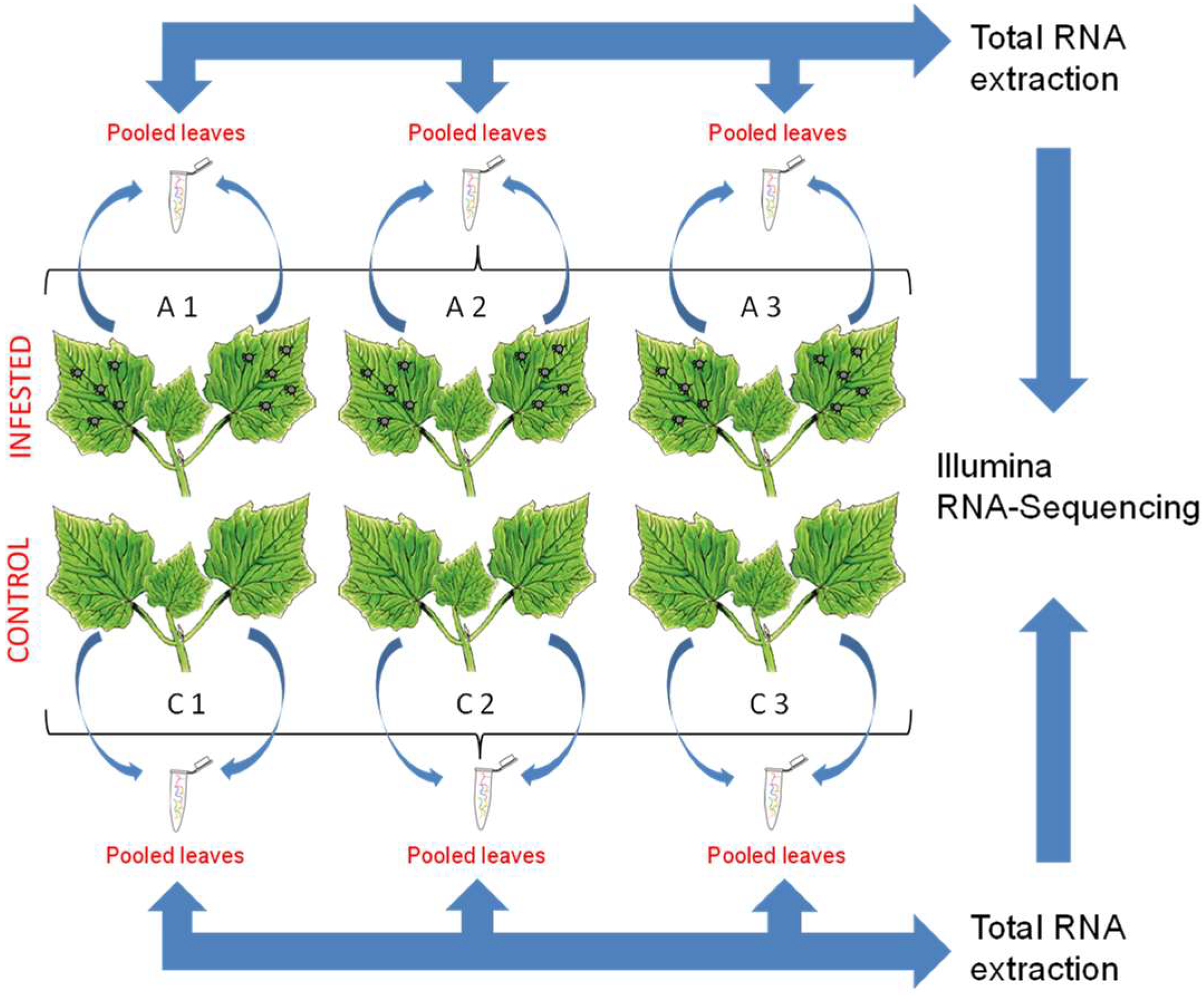
3.1. Biological Material and Experimental Design
3.2. RNA Extraction, Library Construction and Sequencing
3.3. Read Pre-Processing and De Novo Assembly
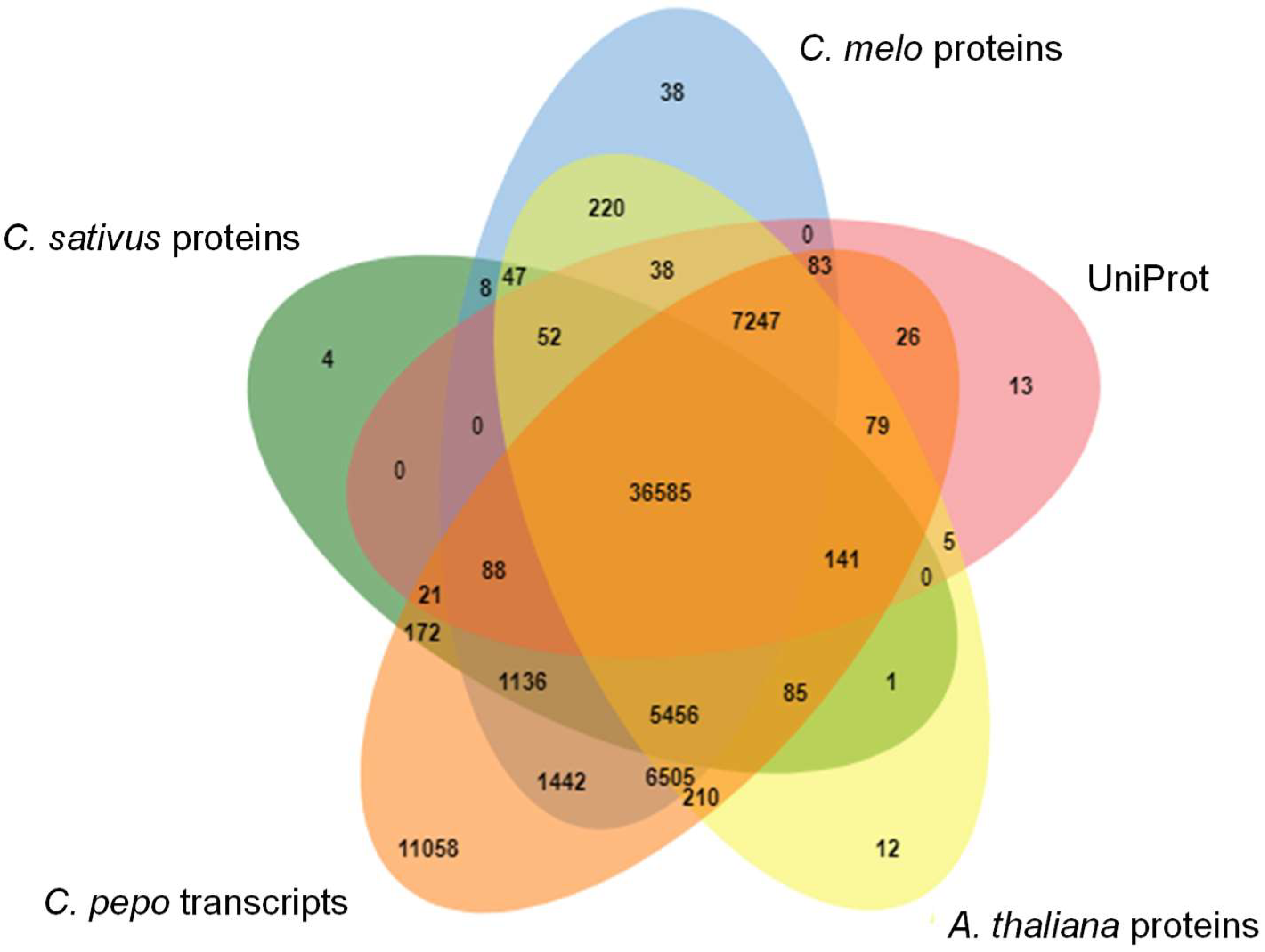
3.4. Transcriptome Annotation
3.5. Mapping Transcripts on the Reference Genome
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Esteras, C.; Gómez, P.; Monforte, A.J.; Blanca, J.; Vicente-Dólera, N.; Roig, C.; Nuez, F.; Picó, B. High-throughput SNP genotyping in Cucurbita pepo for map construction and quantitative trait loci mapping. BMC Genom. 2012, 13, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paris, H.S. Summer squash: History, diversity, and distribution. HortTechnology 1996, 6, 6–13. [Google Scholar]
- Singh, R.; Singh, K. Life history parameters of aphis gossypii glover (homoptera: Aphididae) reared on three vegetable crops. Int. J. Res. Stud. Zool. 2015, 1, 1–9. [Google Scholar]
- Ebert, T.; Cartwright, B. Biology and ecology of aphis gossypii glover (homoptera: Aphididae). Southwest. Entomol. 1997, 22, 116–153. [Google Scholar]
- Blanca, J.; Cañizares, J.; Roig, C.; Ziarsolo, P.; Nuez, F.; Picó, B. Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genom. 2011, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.E.; Strickler, S.R.; Mueller, L.A.; Mazourek, M. An acorn squash (Cucurbita pepo ssp. Ovifera) fruit and seed transcriptome as a resource for the study of fruit traits in cucurbita. Hortic. Res. 2015, 2, 14070. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, A.; Psomopoulos, F.; Ganopoulos, I.; Manioudaki, M.; Tsaftaris, A.; Nianiou-Obeidat, I.; Madesis, P. De novo transcriptome assembly of two contrasting pumpkin cultivars. Genom. Data 2016, 7, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Xanthopoulou, A.; Ganopoulos, I.; Psomopoulos, F.; Manioudaki, M.; Moysiadis, T.; Kapazoglou, A.; Osathanunkul, M.; Michailidou, S.; Kalivas, A.; Tsaftaris, A. De novo comparative transcriptome analysis of genes involved in fruit morphology of pumpkin cultivars with extreme size difference and development of EST-SSR markers. Gene 2017, 622, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, G.; Di Donato, A.; Darrudi, R.; Errico, A.; Aiese Cigliano, R.; Ercolano, M.R. Draft of zucchini (Cucurbita pepo L.) proteome: A resource for genetic and genomic studies. Front. Genet. 2017, 8, 181. [Google Scholar] [CrossRef] [PubMed]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L. De novo assembly of the zucchini genome reveals a whole-genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. Estscan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology, Heidelberg, Germany, 6–10 August 1999; pp. 138–148. [Google Scholar]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Sánchez Alvarado, A.; Yandell, M. Maker: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.; Hannon, G. FASTX-Toolkit: FASTQ/A Short-Reads Preprocessing Tools. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 15 September 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon (Cucumis melo L.). In Proceedings of the National Academy of Sciences of the United States of America, Bethesda, MD, USA, 17 July 2012; pp. 11872–11877. [Google Scholar]
- UniProt Consortium. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Data | High Quality Data | ||
|---|---|---|---|---|
| # Reads | # Paired Reads | # Single Reads | ||
| Control | C24_1 | 31,430,108 | 22,032,822 | 5,256,322 |
| C24_2 | 28,740,043 | 21,066,607 | 4,270,278 | |
| C24_3 | 33,677,909 | 25,020,509 | 4,812,235 | |
| C48_1 | 36,265,357 | 28,423,670 | 4,540,249 | |
| C48_3 | 35,144,118 | 27,591,869 | 4,394,338 | |
| C48_4 | 37,518,763 | 28,808,248 | 5,107,225 | |
| C96_2 | 33,527,557 | 25,873,268 | 4,394,873 | |
| C96_3 | 31,060,525 | 24,146,031 | 4,067,956 | |
| C96_4 | 35,937,098 | 28,576,994 | 4,591,206 | |
| Infested | A24_1 | 35,344,346 | 23,674,612 | 7,451,885 |
| A24_2 | 35,230,308 | 23,732,387 | 7,382,828 | |
| A24_3 | 34,366,050 | 22,964,925 | 7,204,783 | |
| A48_2 | 37,211,641 | 25,261,715 | 7,571,915 | |
| A48_3 | 36,623,056 | 24,901,053 | 7,442,903 | |
| A48_4 | 37,996,974 | 25,666,812 | 7,820,577 | |
| A96_1 | 38,622,935 | 27,596,629 | 5,791,605 | |
| A96_2 | 34,353,147 | 24,532,466 | 5,227,154 | |
| A96_3 | 29,037,507 | 20,856,920 | 4,406,645 | |
| Total # transcripts | 71,648 |
| Total # gene locus | 42,517 |
| # Single sequence | 22,594 |
| # Multiple variants | 19,923 |
| Total sequence length (nt) | 95,354,115 |
| Average transcript length (nt) | 1331 |
| Maximum transcript length (nt) | 12,009 |
| Minimum transcript length (nt) | 100 |
| Median transcript length (nt) | 1084 |
| Items | # Sequences |
|---|---|
| complete ORF | 23,735 |
| 5′ truncated 1 | 25,000 |
| 3′ truncated 2 | 8220 |
| 5′ and 3′ truncated 3 | 10,579 |
| no good ORF | 4114 |
| Total | 71,648 |
| Sample Number | BioSample | SRA ID | Library Name |
|---|---|---|---|
| 1 | SAMN08742104 | SRS3072843 | A24-1 |
| 2 | SAMN08742105 | SRS3072853 | A24-2 |
| 3 | SAMN08742106 | SRS3072846 | A24-3 |
| 4 | SAMN08742107 | SRS3072849 | A48-2 |
| 5 | SAMN08742108 | SRS3072852 | A48-3 |
| 6 | SAMN08742109 | SRS3072855 | A48-4 |
| 7 | SAMN08742110 | SRS3072850 | A96-1 |
| 8 | SAMN08742111 | SRS3072851 | A96-2 |
| 9 | SAMN08742112 | SRS3072848 | A96-3 |
| 10 | SAMN08742113 | SRS3072847 | C24-1 |
| 11 | SAMN08742114 | SRS3072854 | C24-2 |
| 12 | SAMN08742115 | SRS3072842 | C24-3 |
| 13 | SAMN08742116 | SRS3072845 | C48-1 |
| 14 | SAMN08742117 | SRS3072844 | C48-3 |
| 15 | SAMN08742118 | SRS3072839 | C48-4 |
| 16 | SAMN08742119 | SRS3072838 | C96-2 |
| 17 | SAMN08742120 | SRS3072841 | C96-3 |
| 18 | SAMN08742121 | SRS3072840 | C96-4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitiello, A.; Rao, R.; Corrado, G.; Chiaiese, P.; Digilio, M.C.; Cigliano, R.A.; D’Agostino, N. De Novo Transcriptome Assembly of Cucurbita Pepo L. Leaf Tissue Infested by Aphis Gossypii. Data 2018, 3, 36. https://doi.org/10.3390/data3030036
Vitiello A, Rao R, Corrado G, Chiaiese P, Digilio MC, Cigliano RA, D’Agostino N. De Novo Transcriptome Assembly of Cucurbita Pepo L. Leaf Tissue Infested by Aphis Gossypii. Data. 2018; 3(3):36. https://doi.org/10.3390/data3030036
Chicago/Turabian StyleVitiello, Alessia, Rosa Rao, Giandomenico Corrado, Pasquale Chiaiese, Maria Cristina Digilio, Riccardo Aiese Cigliano, and Nunzio D’Agostino. 2018. "De Novo Transcriptome Assembly of Cucurbita Pepo L. Leaf Tissue Infested by Aphis Gossypii" Data 3, no. 3: 36. https://doi.org/10.3390/data3030036
APA StyleVitiello, A., Rao, R., Corrado, G., Chiaiese, P., Digilio, M. C., Cigliano, R. A., & D’Agostino, N. (2018). De Novo Transcriptome Assembly of Cucurbita Pepo L. Leaf Tissue Infested by Aphis Gossypii. Data, 3(3), 36. https://doi.org/10.3390/data3030036