
Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells



{kind=link}
Abstract
:1. Introduction
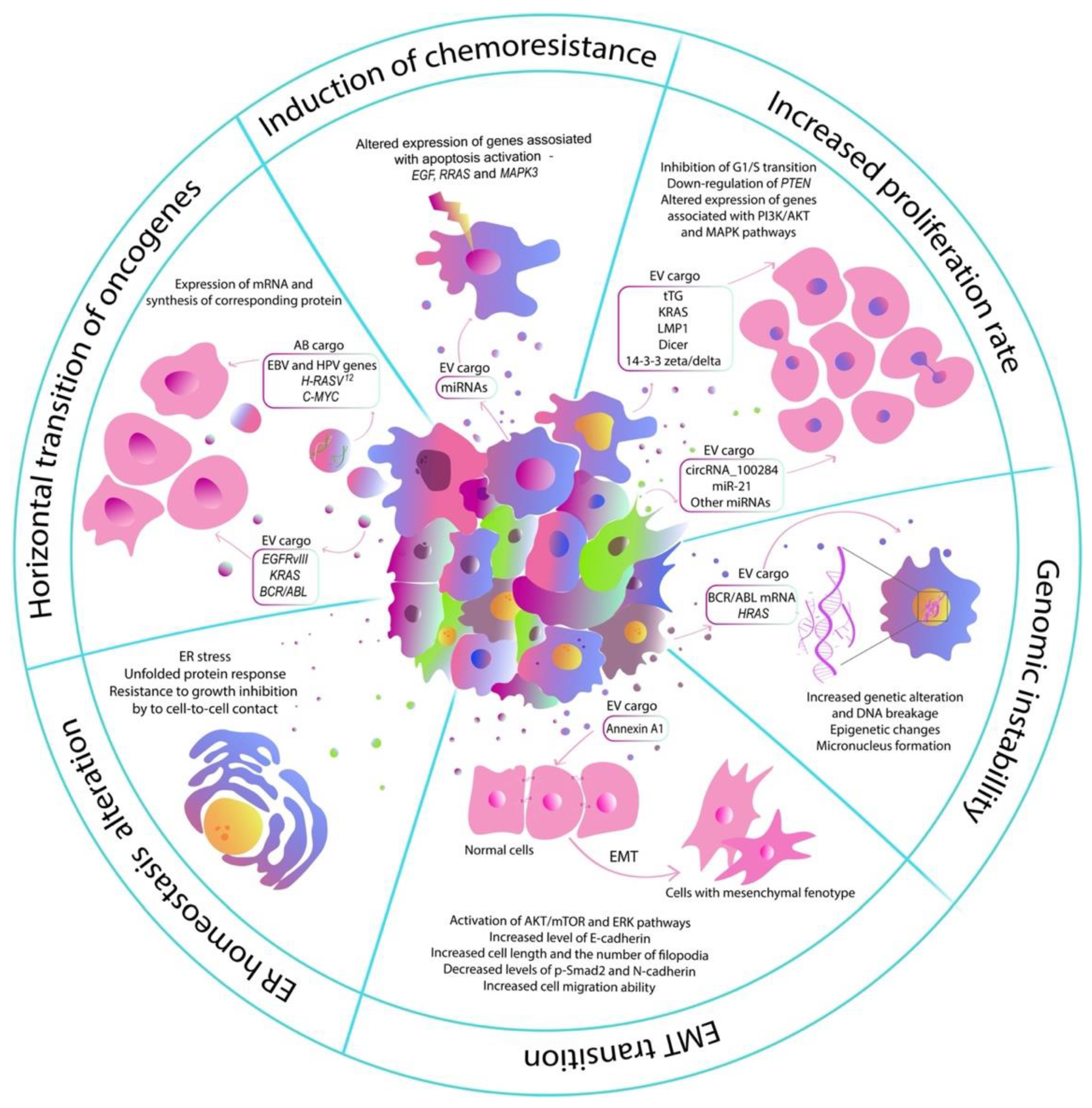
2. Influence of EVs of Tumor Cells on the Normal Cells
2.1. Increased Proliferation Rate
2.2. Induction of Chemoresistance
2.3. Endoplasmic Reticulum Homeostasis Alteration
2.4. Induction of EMT Transition
2.5. Horizontal Transition of Oncogenes
2.6. Induction of Genomic Instability
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Parsa, N. Environmental factors inducing human cancers. Iran. J. Public Health 2012, 41, 1–9. [Google Scholar] [PubMed]
- Mertz, T.M.; Harcy, V.; Roberts, S.A. Risks at the DNA Replication Fork: Effects upon Carcinogenesis and Tumor Heterogeneity. Genes 2017, 8, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.X.; Liu, X.M.; Lv, M.M.; Chen, L.; Zhao, J.H.; Zhong, S.L.; Ji, M.H.; Hu, Q.; Luo, Z.; Wu, J.Z.; et al. Exosomes from Drug-Resistant Breast Cancer Cells Transmit Chemoresistance by a Horizontal Transfer of MicroRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef]
- Li, S.; Zhou, J.; Wu, H.; Lu, Q.; Tai, Y.; Liu, Q.; Wang, C. Oncogenic transformation of normal breast epithelial cells co-cultured with cancer cells. Cell Cycle 2018, 17, 2027–2040. [Google Scholar] [CrossRef] [Green Version]
- Mittra, I.; Samant, U.; Sharma, S.; Raghuram, G.V.; Saha, T.; Tidke, P.; Pancholi, N.; Gupta, D.; Prasannan, P.; Gaikwad, A.; et al. Cell-free chromatin from dying cancer cells integrate into genomes of bystander healthy cells to induce DNA damage and inflammation. Cell Death Discov. 2017, 3, 17015. [Google Scholar] [CrossRef] [Green Version]
- Souza, A.G.; Bastos, V.A.F.; Fujimura, P.T.; Ferreira, I.C.C.; Leal, L.F.; da Silva, L.S.; Laus, A.C.; Reis, R.M.; Martins, M.M.; Santos, P.S.; et al. Cell-free DNA promotes malignant transformation in non-tumor cells. Sci. Rep. 2020, 10, 21674. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Gilazieva, Z.E.; Kletukhina, S.K.; Aimaletdinov, A.M.; Garanina, E.E.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Cytochalasin B-Induced Membrane Vesicles from Human Mesenchymal Stem Cells Overexpressing IL2 Are Able to Stimulate CD8(+) T-Killers to Kill Human Triple Negative Breast Cancer Cells. Biology 2021, 10, 141. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic Prospects of Extracellular Vesicles in Cancer Treatment. Front. Immunol 2018, 9, 1534. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Raimondo, S.; Pucci, M.; Alessandro, R.; Fontana, S. Extracellular Vesicles and Tumor-Immune Escape: Biological Functions and Clinical Perspectives. Int. J. Mol. Sci. 2020, 21, 2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhang, P.; Wang, Y.; Wang, J.; Su, M.; Wang, Y.; Zhou, L.; Zhou, J.; Xiong, W.; Zeng, Z.; et al. The Biogenesis, Biology, and Clinical Significance of Exosomal PD-L1 in Cancer. Front. Immunol. 2020, 11, 604. [Google Scholar] [CrossRef] [PubMed]
- Kogure, A.; Yoshioka, Y.; Ochiya, T. Extracellular Vesicles in Cancer Metastasis: Potential as Therapeutic Targets and Materials. Int. J. Mol. Sci. 2020, 21, 4463. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Gangadaran, P.; Ahn, B.C. A novel strategy of transferring NIS protein to cells using extracellular vesicles leads to increase in iodine uptake and cytotoxicity. Int J. Nanomed 2019, 14, 1779–1787. [Google Scholar] [CrossRef] [Green Version]
- Beckler, M.D.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic Analysis of Exosomes from Mutant KRAS Colon Cancer Cells Identifies Intercellular Transfer of Mutant KRAS. Mol. Cell. Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Ji, X.; Liu, J.; Fan, D.; Zhou, Q.; Chen, C.; Wang, W.; Wang, G.; Wang, H.; Yuan, W.; et al. Effects of exosomes on pre-metastatic niche formation in tumors. Mol. Cancer 2019, 18, 39. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Marangoni, K.; Bastos, V.A.F.; Alves, P.T.; Goulart, L.R.; Alonso-Goulart, V. Extracellular vesicles as drivers of epithelial-mesenchymal transition and carcinogenic characteristics in normal prostate cells. Mol. Carcinog. 2018, 57, 503–511. [Google Scholar] [CrossRef]
- Ozawa, P.M.M.; Alkhilaiwi, F.; Cavalli, I.J.; Malheiros, D.; Ribeiro, E.M.D.F.; Cavalli, L.R. Extracellular vesicles from triple-negative breast cancer cells promote proliferation and drug resistance in non-tumorigenic breast cells. Breast Cancer Res. Treat. 2018, 172, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; You, Y.; Li, Q.; Zeng, C.; Fu, F.; Guo, A.; Zhang, H.; Zou, P.; Zhong, Z.; Wang, H.; et al. BCR-ABL1-positive microvesicles transform normal hematopoietic transplants through genomic instability: Implications for donor cell leukemia. Leukemia 2014, 28, 1666–1675. [Google Scholar] [CrossRef]
- Mulvey, H.E.; Chang, A.; Adler, J.; Del Tatto, M.; Perez, K.; Quesenberry, P.J.; Chatterjee, D. Extracellular vesicle-mediated phenotype switching in malignant and non-malignant colon cells. BMC Cancer 2015, 15, 571. [Google Scholar] [CrossRef]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J. Immunol. 2019, 203, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.Y.; Chen, C.; Yang, Q.L.; Xue, J.C.; Chen, X.; Sun, B.F.; Luo, F.; Liu, X.L.; Xiao, T.; Xu, H.; et al. Exosomal circRNA_100284 from arsenite-transformed cells, via microRNA-217 regulation of EZH2, is involved in the malignant transformation of human hepatic cells by accelerating the cell cycle and promoting cell proliferation. Cell Death Dis. 2018, 9, 454. [Google Scholar] [CrossRef]
- Xu, Y.; Luo, F.; Liu, Y.; Shi, L.; Lu, X.L.; Xu, W.C.; Liu, Q.Z. Exosomal miR-21 derived from arsenite-transformed human bronchial epithelial cells promotes cell proliferation associated with arsenite carcinogenesis. Arch. Toxicol. 2015, 89, 1071–1082. [Google Scholar] [CrossRef]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer Exosomes Perform Cell-Independent MicroRNA Biogenesis and Promote Tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, K.; Cross-Knorr, S.; Dillard, C.; Pantazatos, D.; Del Tatto, M.; Mills, D.; Goldstein, L.; Renzulli, J.; Quesenberry, P.; Chatterjee, D. Reversal of chemosensitivity and induction of cell malignancy of a non-malignant prostate cancer cell line upon extracellular vesicle exposure. Mol. Cancer 2013, 12, 118. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.X.; Ding, C.; Yu, S.Q. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Silvers, C.R.; Messing, E.M.; Lee, Y.F. Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells. J. Biol. Chem. 2019, 294, 3207–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Carbayo, M.; Socci, N.D.; Lozano, J.; Saint, F.; Cordon-Cardo, C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 2006, 24, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Bychkov, M.L.; Kirichenko, A.V.; Mikhaylova, I.N.; Paramonov, A.S.; Yastremsky, E.V.; Kirpichnikov, M.P.; Shulepko, M.A.; Lyukmanova, E.N. Extracellular Vesicles Derived from Acidified Metastatic Melanoma Cells Stimulate Growth, Migration, and Stemness of Normal Keratinocytes. Biomedicines 2022, 10, 660. [Google Scholar] [CrossRef]
- Wang, S.W.; Ju, T.Y.; Wang, J.J.; Yang, F.; Qu, K.G.; Liu, W.; Wang, Z.B. Migration of BEAS-2B cells enhanced by H1299 cell derived-exosomes. Micron 2021, 143, 103001. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.C.; Liu, W.; Wang, Z.L.; Wang, C.; Ai, Z.L. Exosomal ANXA1 derived from thyroid cancer cells is associated with malignant transformation of human thyroid follicular epithelial cells by promoting cell proliferation. Int. J. Oncol. 2021, 59, 104. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFrvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Garcia-Olmo, D.C.; Dominguez, C.; Garcia-Arranz, M.; Anker, P.; Stroun, M.; Garcia-Verdugo, J.M.; Garcia-Olmo, D. Cell-Free Nucleic Acids Circulating in the Plasma of Colorectal Cancer Patients Induce the Oncogenic Transformation of Susceptible Cultured Cells. Cancer Res. 2010, 70, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Han, Y.; Ren, H.M.; Chen, C.Y.; He, D.F.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C.Y. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wu, G.Z.; Tan, X.R.; Han, Y.; Chen, C.Y.; Li, C.W.; Wang, N.; Zou, X.; Chen, X.J.; Zhou, F.Y.; et al. Transferred BCR/ABL DNA from K562 Extracellular Vesicles Causes Chronic Myeloid Leukemia in Immunodeficient Mice. PLoS ONE 2014, 9, e105200. [Google Scholar] [CrossRef] [Green Version]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.Y.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermetet, F.; Jacquin, E.; Launay, S.; Gaiffe, E.; Couturier, M.; Hirchaud, F.; Sandoz, P.; Pretet, J.L.; Mougin, C. Efferocytosis of apoptotic human papillomavirus-positive cervical cancer cells by human primary fibroblasts. Biol. Cell 2016, 108, 189–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaiffe, E.; Pretet, J.L.; Launay, S.; Jacquin, E.; Saunier, M.; Hetzel, G.; Oudet, P.; Mougin, C. Apoptotic HPV Positive Cancer Cells Exhibit Transforming Properties. PLoS ONE 2012, 7, e36766. [Google Scholar] [CrossRef]
- Holmgren, L.; Szeles, A.; Rajnavolgyi, E.; Folkman, J.; Klein, G.; Ernberg, I.; Falk, K.I. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood 1999, 93, 3956–3963. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Chennakrishnaiah, S.; Tsering, T.; Gregory, C.; Tawil, N.; Spinelli, C.; Montermini, L.; Karatzas, N.; Aprikian, S.; Choi, D.; Klewes, L.; et al. Extracellular vesicles from genetically unstable, oncogene-driven cancer cells trigger micronuclei formation in endothelial cells. Sci. Rep. 2020, 10, 8532. [Google Scholar] [CrossRef]
- Li, X.; Gheinani, A.H.; Adam, R.M. A multi-omics approach to understanding the field effect in bladder cancer. Transl. Urol. 2019, 8, 775–778. [Google Scholar] [CrossRef]
- Hayatudin, R.; Fong, Z.; Ming, L.C.; Goh, B.H.; Lee, W.L.; Kifli, N. Overcoming Chemoresistance via Extracellular Vesicle Inhibition. Front. Mol. BioSci. 2021, 8, 629874. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulpanova, D.S.; Pukhalskaia, T.V.; Rizvanov, A.A.; Solovyeva, V.V. Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells. Bioengineering 2022, 9, 245. https://doi.org/10.3390/bioengineering9060245
Chulpanova DS, Pukhalskaia TV, Rizvanov AA, Solovyeva VV. Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells. Bioengineering. 2022; 9(6):245. https://doi.org/10.3390/bioengineering9060245
Chicago/Turabian StyleChulpanova, Daria S., Tamara V. Pukhalskaia, Albert A. Rizvanov, and Valeriya V. Solovyeva. 2022. "Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells" Bioengineering 9, no. 6: 245. https://doi.org/10.3390/bioengineering9060245
APA StyleChulpanova, D. S., Pukhalskaia, T. V., Rizvanov, A. A., & Solovyeva, V. V. (2022). Contribution of Tumor-Derived Extracellular Vesicles to Malignant Transformation of Normal Cells. Bioengineering, 9(6), 245. https://doi.org/10.3390/bioengineering9060245