
Present Application and Perspectives of Organoid Imaging Technology



Abstract
:1. Introduction
2. Imaging Technology
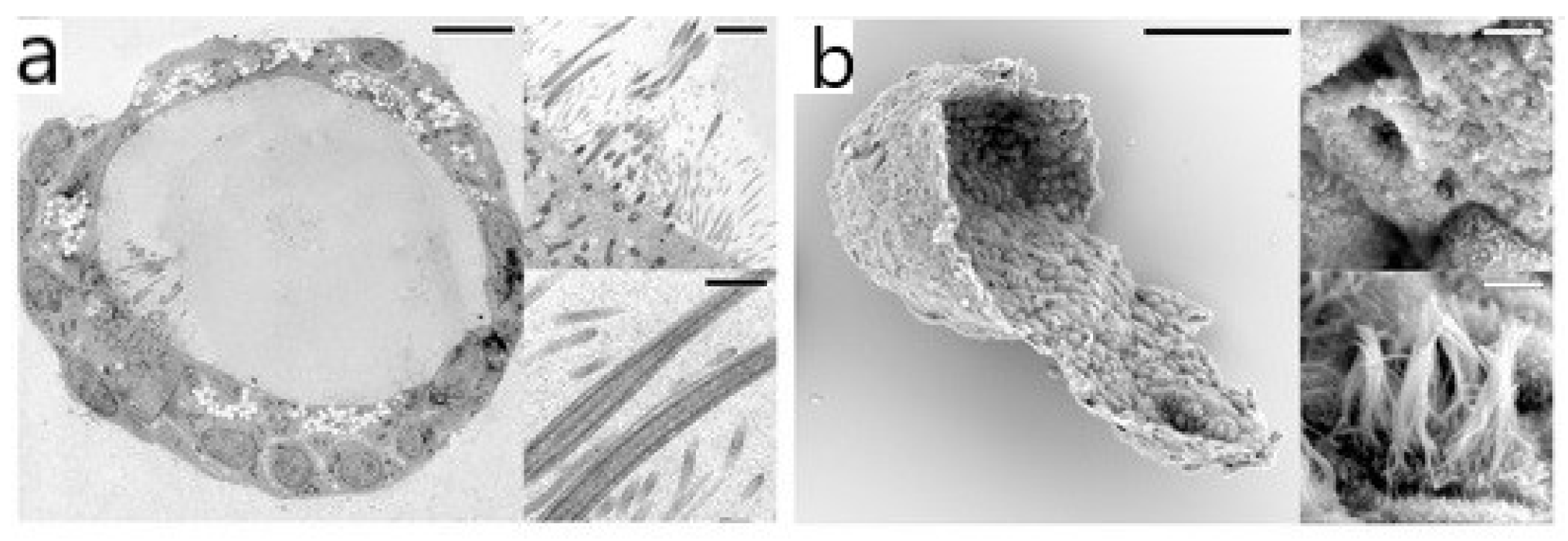
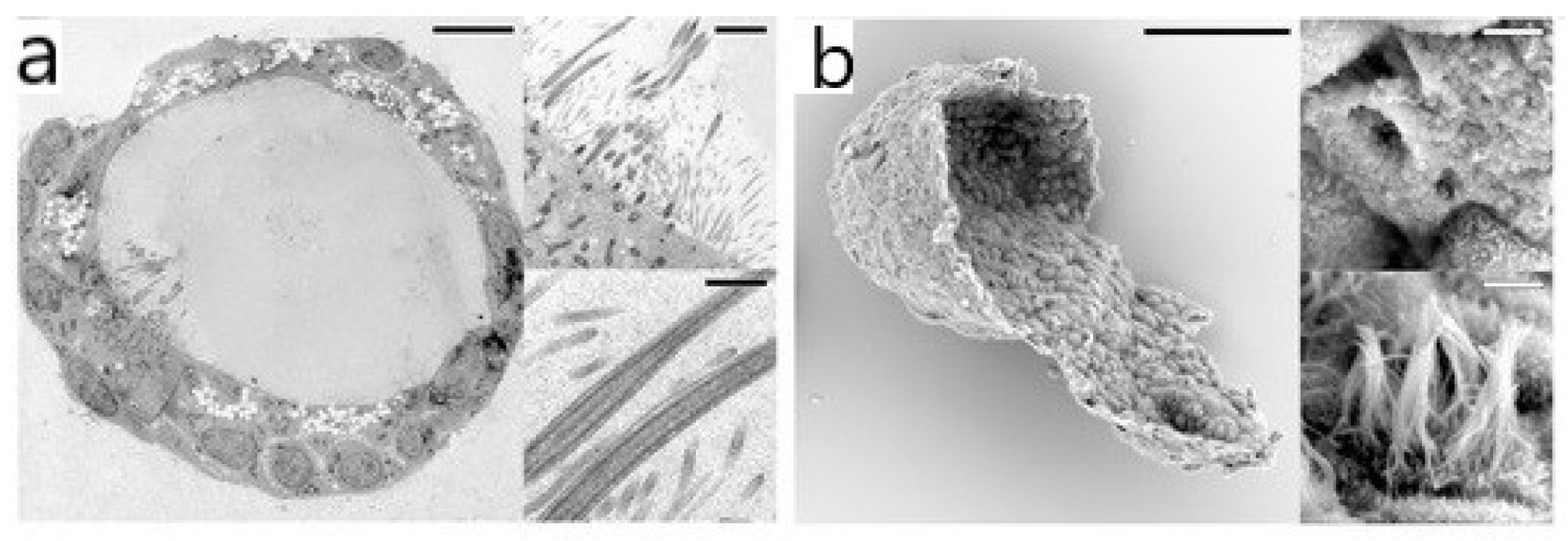
2.1. Electron Microscopy (EM)
2.2. Bright-Field Microscopy
2.3. Fluorescence Microscopy
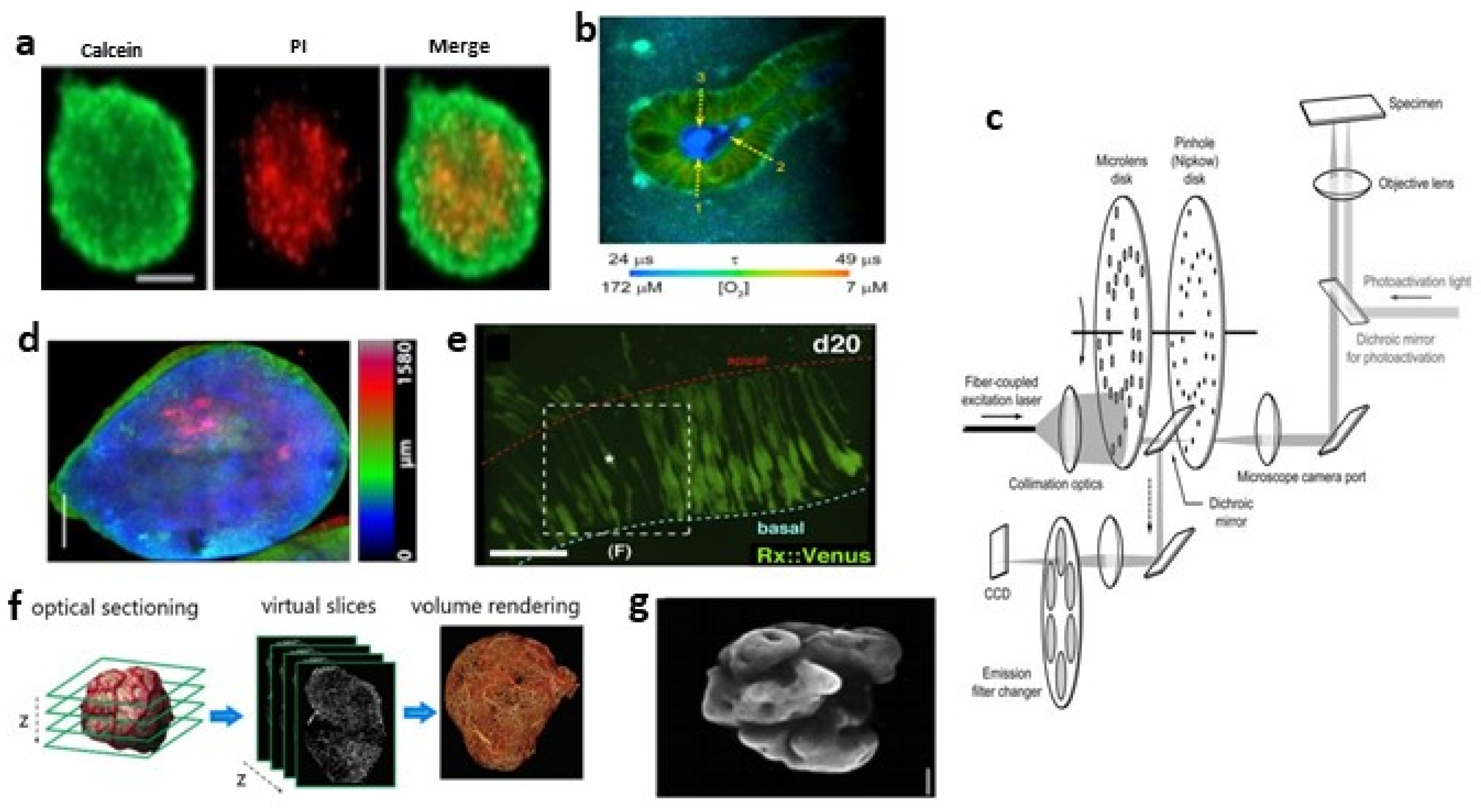
2.3.1. Wide-Field Fluorescence Microscopy (WFFM)
2.3.2. Laser Scanning Confocal Microscopy (LSCM)
2.3.3. Spinning Disc Confocal (SDC) Microscopy
2.3.4. Multiphoton Microscopy
2.3.5. Light Sheet Fluorescence Microscopy (LSFM)
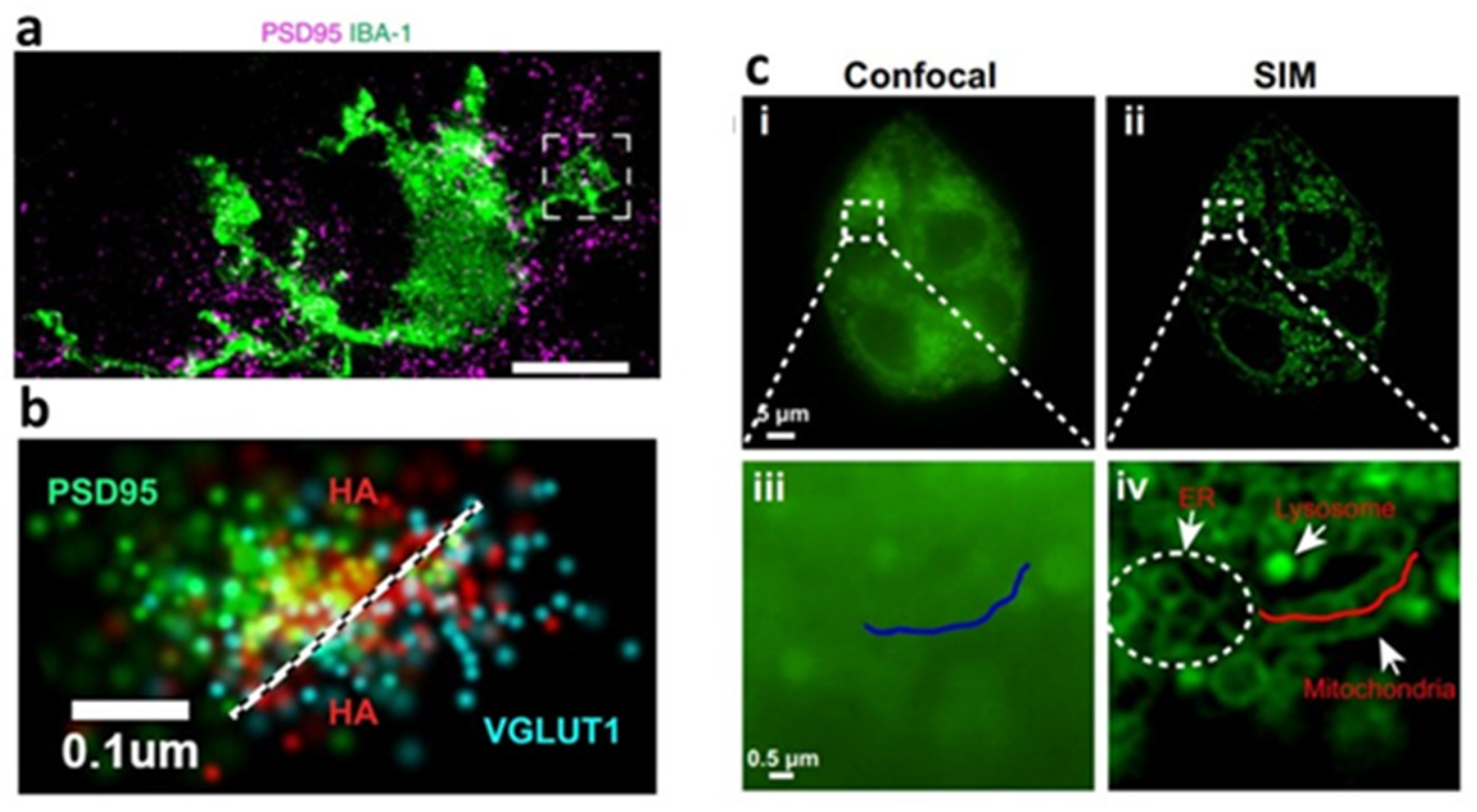
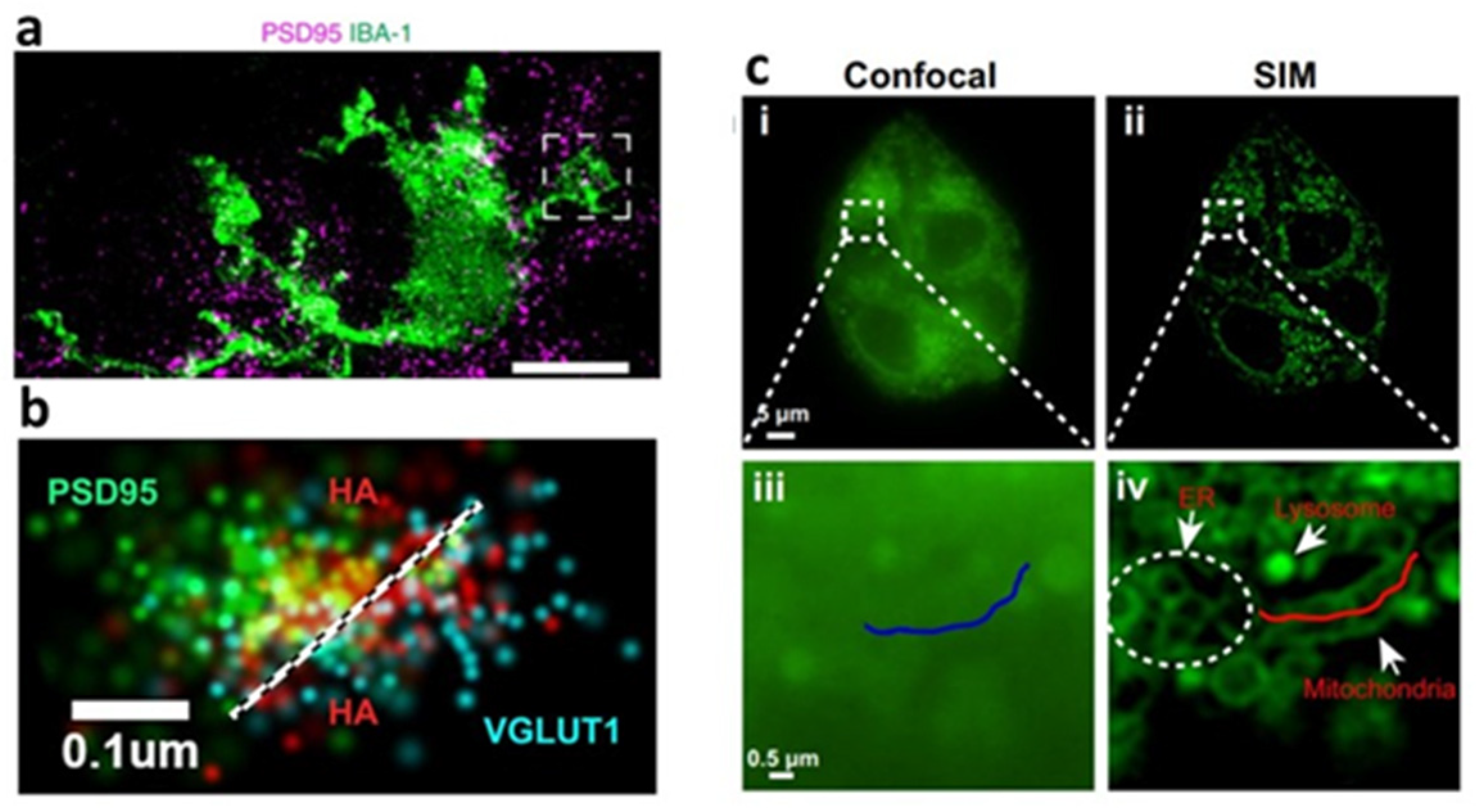
2.3.6. Super-Resolution Fluorescence Microscopy
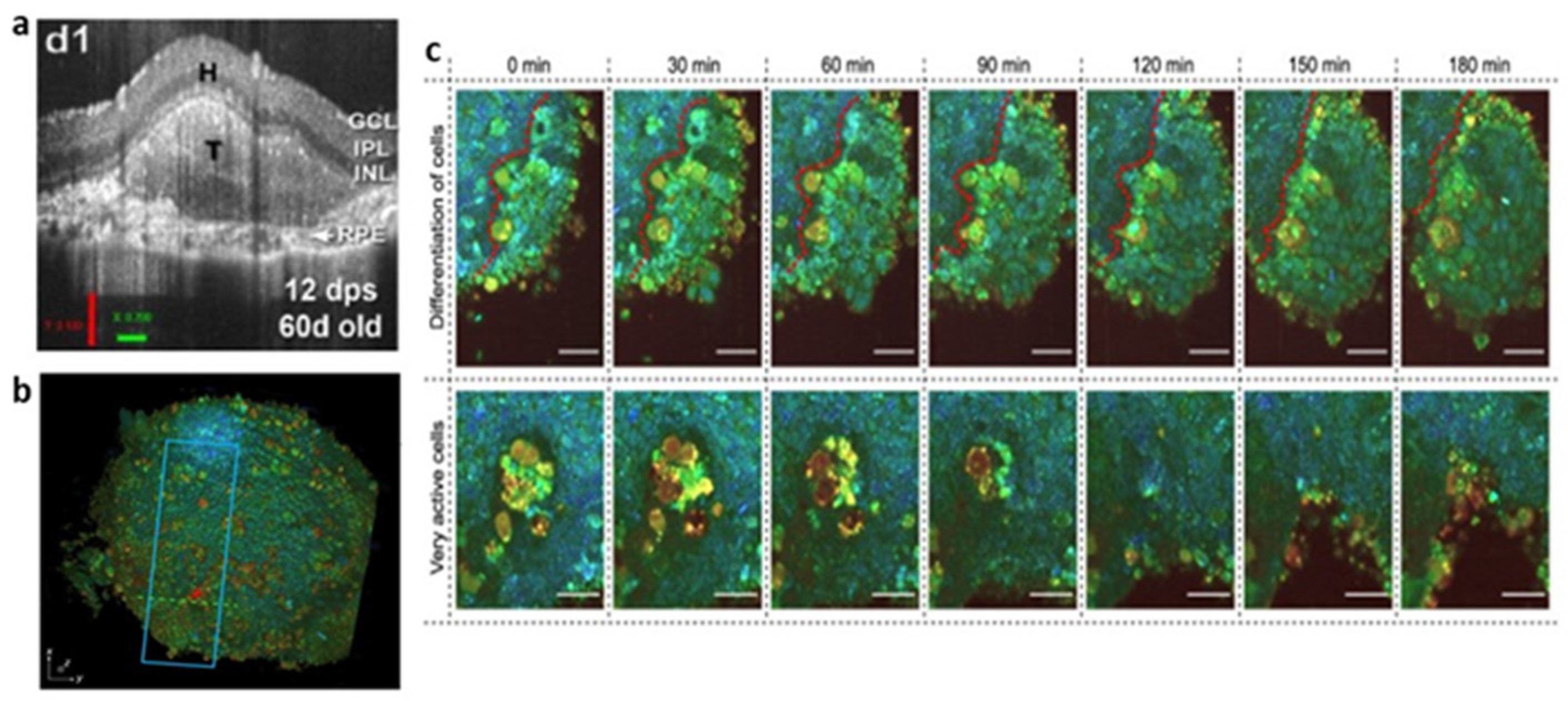
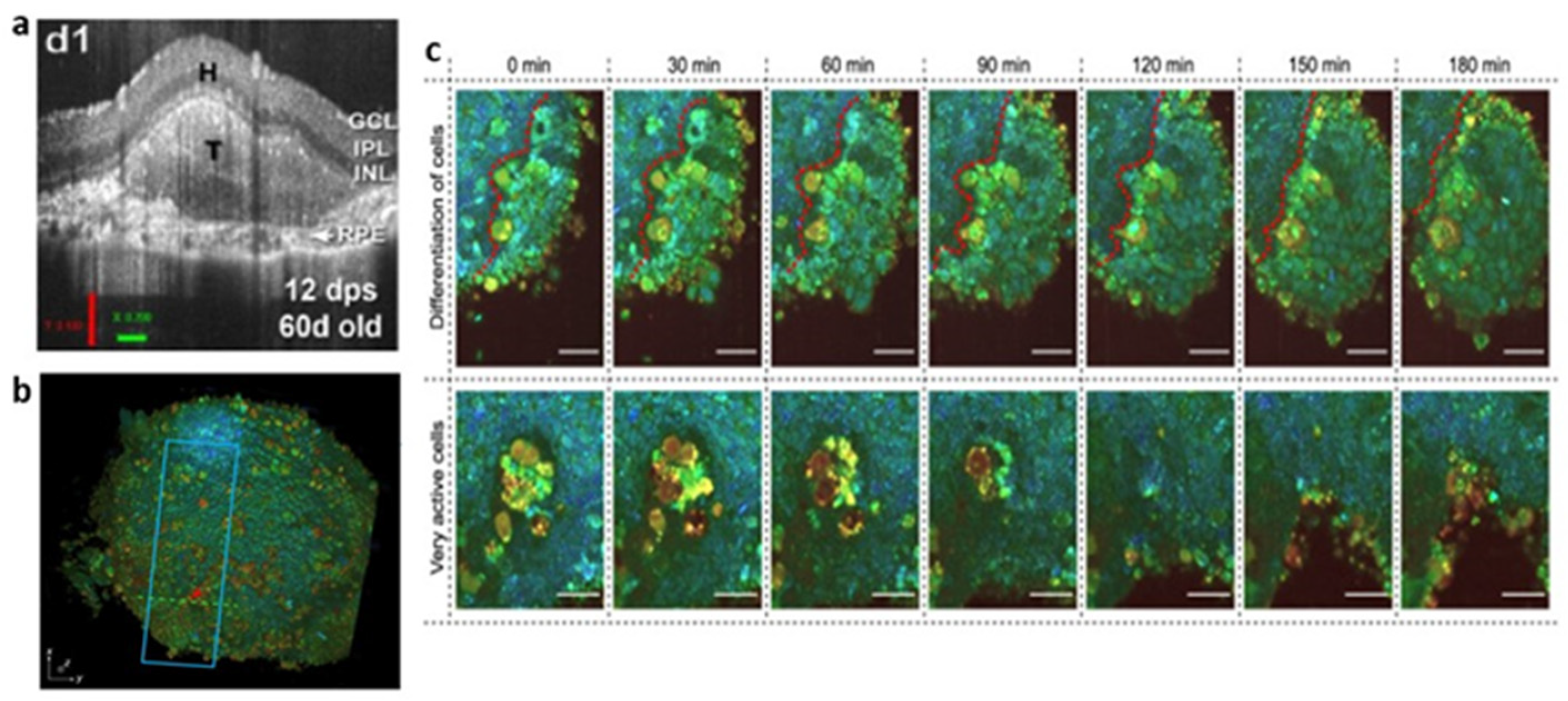
2.4. Optical Coherence Tomography (OCT)
2.5. Others
3. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an In Vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.H.; Nattiv, R.; Dedhia, P.H.; Nagy, M.S.; Chin, A.M.; Thomson, M.; Klein, O.D.; Spence, J.R. In Vitro patterning of pluripotent stem cell-derived intestine recapitulates In Vivo human development. Development 2017, 144, 1045–1055. [Google Scholar] [CrossRef] [Green Version]
- Roper, J.; Tammela, T.; Akkad, A.; Almeqdadi, M.; Santos, S.B.; Jacks, T.; Yilmaz, O.H. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc. 2018, 13, 217–234. [Google Scholar] [CrossRef]
- McCracken, K.W.; Cata, E.M.; Crawford, C.M.; Sinagoga, K.L.; Schumacher, M.; Rockich, B.E.; Tsai, Y.H.; Mayhew, C.N.; Spence, J.R.; Zavros, Y.; et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 2014, 516, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.K.; Ninomiya, N.; Sekine, M.; Komazaki, S.; Wang, P.C.; Asashima, M.; Kurisaki, A. Generation of stomach tissue from mouse embryonic stem cells. Nat. Cell Biol. 2015, 17, 984–993. [Google Scholar] [CrossRef]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef]
- Taguchi, A.; Nishinakamura, R. Higher-Order Kidney Organogenesis from Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 730–746.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [Green Version]
- Bagley, J.A.; Reumann, D.; Bian, S.; Levi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Volkner, M.; Zschatzsch, M.; Rostovskaya, M.; Overall, R.W.; Busskamp, V.; Anastassiadis, K.; Karl, M.O. Retinal Organoids from Pluripotent Stem Cells Efficiently Recapitulate Retinogenesis. Stem Cell Rep. 2016, 6, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2019, 146, dev171686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Hohwieler, M.; Illing, A.; Hermann, P.C.; Mayer, T.; Stockmann, M.; Perkhofer, L.; Eiseler, T.; Antony, J.S.; Muller, M.; Renz, S.; et al. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut 2017, 66, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Jarde, T.; Lloyd-Lewis, B.; Thomas, M.; Kendrick, H.; Melchor, L.; Bougaret, L.; Watson, P.D.; Ewan, K.; Smalley, M.J.; Dale, T.C. Wnt and Neuregulin1/ErbB signalling extends 3D culture of hormone responsive mammary organoids. Nat. Commun. 2016, 7, 13207. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, P.R.; Dekkers, J.F.; Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. Derivation of a robust mouse mammary organoid system for studying tissue dynamics. Development 2017, 144, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [Green Version]
- Sampaziotis, F.; de Brito, M.C.; Madrigal, P.; Bertero, A.; Saeb-Parsy, K.; Soares, F.A.C.; Schrumpf, E.; Melum, E.; Karlsen, T.H.; Bradley, J.A.; et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat. Biotechnol. 2015, 33, 845–852. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [Green Version]
- Kurmann, A.A.; Serra, M.; Hawkins, F.; Rankin, S.A.; Mori, M.; Astapova, I.; Ullas, S.; Lin, S.; Bilodeau, M.; Rossant, J.; et al. Regeneration of Thyroid Function by Transplantation of Differentiated Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 527–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonica, F.; Kasprzyk, D.F.; Schiavo, A.A.; Romitti, M.; Costagliola, S. Generation of Functional Thyroid Tissue Using 3D-Based Culture of Embryonic Stem Cells. Methods Mol. Biol. 2017, 1597, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Maimets, M.; Rocchi, C.; Bron, R.; Pringle, S.; Kuipers, J.; Giepmans, B.N.; Vries, R.G.; Clevers, H.; de Haan, G.; van Os, R.; et al. Long-Term In Vitro Expansion of Salivary Gland Stem Cells Driven by Wnt Signals. Stem Cell Rep. 2016, 6, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.S.; Lee, S.; Hong, H.J.; Lim, Y.C.; Koh, W.G.; Lim, J.Y. Stem cell properties of human clonal salivary gland stem cells are enhanced by three-dimensional priming culture in nanofibrous microwells. Stem Cell Res. Ther. 2018, 9, 74. [Google Scholar] [CrossRef]
- Turco, M.Y.; Gardner, L.; Hughes, J.; Cindrova-Davies, T.; Gomez, M.J.; Farrell, L.; Hollinshead, M.; Marsh, S.G.E.; Brosens, J.J.; Critchley, H.O.; et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 2017, 19, 568–577. [Google Scholar] [CrossRef]
- Fitzgerald, H.C.; Dhakal, P.; Behura, S.K.; Schust, D.J.; Spencer, T.E. Self-renewing endometrial epithelial organoids of the human uterus. Proc. Natl. Acad. Sci. USA 2019, 116, 23132–23142. [Google Scholar] [CrossRef]
- Ren, W.; Lewandowski, B.C.; Watson, J.; Aihara, E.; Iwatsuki, K.; Bachmanov, A.A.; Margolskee, R.F.; Jiang, P. Single Lgr5- or Lgr6-expressing taste stem/progenitor cells generate taste bud cells ex vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 16401–16406. [Google Scholar] [CrossRef] [Green Version]
- Aihara, E.; Mahe, M.M.; Schumacher, M.A.; Matthis, A.L.; Feng, R.; Ren, W.; Noah, T.K.; Matsu-ura, T.; Moore, S.R.; Hong, C.I.; et al. Characterization of stem/progenitor cell cycle using murine circumvallate papilla taste bud organoid. Sci. Rep. 2015, 5, 17185. [Google Scholar] [CrossRef] [Green Version]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In Vitro generation of human pluripotent stem cell derived lung organoids. eLife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Bottinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Lichtman, J.W. SnapShot: Tissue Clearing. Cell 2017, 171, 496–496.e1. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Jamieson, P.R.; Pal, B.; Whitehead, L.; Nicholas, K.R.; Lindeman, G.J.; Visvader, J.E. Essential role for a novel population of binucleated mammary epithelial cells in lactation. Nat. Commun. 2016, 7, 11400. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Alieva, M.; Wellens, L.M.; Ariese, H.C.R.; Jamieson, P.R.; Vonk, A.M.; Amatngalim, G.D.; Hu, H.; Oost, K.C.; Snippert, H.J.G.; et al. High-resolution 3D imaging of fixed and cleared organoids. Nat. Protoc. 2019, 14, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., III; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [Green Version]
- Bremond Martin, C.; Simon Chane, C.; Clouchoux, C.; Histace, A. Recent Trends and Perspectives in Cerebral Organoids Imaging and Analysis. Front. Neurosci. 2021, 15, 629067. [Google Scholar] [CrossRef]
- Zewail, A.H. Four-dimensional electron microscopy. Science 2010, 328, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. eLife 2019, 8, e50333. [Google Scholar] [CrossRef]
- Gomez-Giro, G.; Arias-Fuenzalida, J.; Jarazo, J.; Zeuschner, D.; Ali, M.; Possemis, N.; Bolognin, S.; Halder, R.; Jager, C.; Kuper, W.F.E.; et al. Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2019, 7, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovando-Roche, P.; West, E.L.; Branch, M.J.; Sampson, R.D.; Fernando, M.; Munro, P.; Georgiadis, A.; Rizzi, M.; Kloc, M.; Naeem, A.; et al. Use of bioreactors for culturing human retinal organoids improves photoreceptor yields. Stem Cell Res. Ther. 2018, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.N.T.; Harbison, A.M. Scanning Electron Microscopy Sample Preparation and Imaging. Methods Mol. Biol. 2017, 1606, 71–84. [Google Scholar] [CrossRef]
- Schutgens, F.; Rookmaaker, M.B.; Margaritis, T.; Rios, A.; Ammerlaan, C.; Jansen, J.; Gijzen, L.; Vormann, M.; Vonk, A.; Viveen, M.; et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019, 37, 303–313. [Google Scholar] [CrossRef]
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I.; et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Rep. 2017, 8, 1144–1154. [Google Scholar] [CrossRef]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brandl, B.; Muller, F.J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Borten, M.A.; Bajikar, S.S.; Sasaki, N.; Clevers, H.; Janes, K.A. Automated brightfield morphometry of 3D organoid populations by OrganoSeg. Sci. Rep. 2018, 8, 5319. [Google Scholar] [CrossRef] [Green Version]
- Kassis, T.; Hernandez-Gordillo, V.; Langer, R.; Griffith, L.G. OrgaQuant: Human Intestinal Organoid Localization and Quantification Using Deep Convolutional Neural Networks. Sci. Rep. 2019, 9, 12479. [Google Scholar] [CrossRef] [Green Version]
- Hasnain, Z.; Fraser, A.K.; Georgess, D.; Choi, A.; Macklin, P.; Bader, J.S.; Peyton, S.R.; Ewald, A.J.; Newton, P.K. OrgDyn: Feature- and model-based characterization of spatial and temporal organoid dynamics. Bioinformatics 2020, 36, 3292–3294. [Google Scholar] [CrossRef]
- Sartore, R.C.; Cardoso, S.C.; Lages, Y.V.; Paraguassu, J.M.; Stelling, M.P.; Madeiro da Costa, R.F.; Guimaraes, M.Z.; Perez, C.A.; Rehen, S.K. Trace elements during primordial plexiform network formation in human cerebral organoids. PeerJ 2017, 5, e2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivitilli, A.A.; Gosio, J.T.; Ghoshal, B.; Evstratova, A.; Trcka, D.; Ghiasi, P.; Hernandez, J.J.; Beaulieu, J.M.; Wrana, J.L.; Attisano, L. Robust production of uniform human cerebral organoids from pluripotent stem cells. Life Sci. Alliance 2020, 3, e202000707. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, I.Y.; Kielkowski, P.; Giorgio, G.; O’Neill, A.C.; Di Giaimo, R.; Kyrousi, C.; Khattak, S.; Sieber, S.A.; Robertson, S.P.; Cappello, S. ECE2 regulates neurogenesis and neuronal migration during human cortical development. EMBO Rep. 2020, 21, e48204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.F. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, M.J.; Smith, I.; Parker, I.; Bootman, M.D. Fluorescence microscopy. Cold Spring Harb. Protoc. 2014, 2014, pdb-top071795. [Google Scholar] [CrossRef] [Green Version]
- Piston, D.W.; Knobel, S.M. Real-time Analysis of Glucose Metabolism by Microscopy. Trends Endocrinol. Metab. 1999, 10, 413–417. [Google Scholar] [CrossRef]
- Hoebe, R.A.; Van Oven, C.H.; Gadella, T.W., Jr.; Dhonukshe, P.B.; Van Noorden, C.J.; Manders, E.M. Controlled light-exposure microscopy reduces photobleaching and phototoxicity in fluorescence live-cell imaging. Nat. Biotechnol. 2007, 25, 249–253. [Google Scholar] [CrossRef]
- Frigault, M.M.; Lacoste, J.; Swift, J.L.; Brown, C.M. Live-cell microscopy-tips and tools. J. Cell Sci. 2009, 122, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Weber, P.; Bruns, T.; Strauss, W.S.; Wittig, R.; Schneckenburger, H. Light dose is a limiting factor to maintain cell viability in fluorescence microscopy and single molecule detection. Int. J. Mol. Sci. 2010, 11, 956–966. [Google Scholar] [CrossRef] [Green Version]
- Bernas, T.; Zarebski, M.; Dobrucki, J.W.; Cook, P.R. Minimizing photobleaching during confocal microscopy of fluorescent probes bound to chromatin: Role of anoxia and photon flux. J. Microsc. 2004, 215, 281–296. [Google Scholar] [CrossRef]
- Vrouenraets, M.B.; Visser, G.W.; Snow, G.B.; van Dongen, G.A. Basic principles, applications in oncology and improved selectivity of photodynamic therapy. Anticancer Res. 2003, 23, 505–522. [Google Scholar] [PubMed]
- Dixit, R.; Cyr, R. Cell damage and reactive oxygen species production induced by fluorescence microscopy: Effect on mitosis and guidelines for non-invasive fluorescence microscopy. Plant J. 2003, 36, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icha, J.; Weber, M.; Waters, J.C.; Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. Bioessays 2017, 39, 1700003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiepas, A.; Voorand, E.; Mubaid, F.; Siegel, P.M.; Brown, C.M. Optimizing live-cell fluorescence imaging conditions to minimize phototoxicity. J. Cell Sci. 2020, 133, jcs242834. [Google Scholar] [CrossRef]
- Ettinger, A.; Wittmann, T. Fluorescence live cell imaging. Methods Cell Biol. 2014, 123, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Agard, D.A.; Hiraoka, Y.; Shaw, P.; Sedat, J.W. Fluorescence microscopy in three dimensions. Methods Cell Biol. 1989, 30, 353–377. [Google Scholar] [CrossRef]
- Antonica, F.; Kasprzyk, D.F.; Opitz, R.; Iacovino, M.; Liao, X.H.; Dumitrescu, A.M.; Refetoff, S.; Peremans, K.; Manto, M.; Kyba, M.; et al. Generation of functional thyroid from embryonic stem cells. Nature 2012, 491, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Dye, B.R.; Dedhia, P.H.; Miller, A.J.; Nagy, M.S.; White, E.S.; Shea, L.D.; Spence, J.R. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. eLife 2016, 5, e19732. [Google Scholar] [CrossRef]
- Linnemann, J.R.; Miura, H.; Meixner, L.K.; Irmler, M.; Kloos, U.J.; Hirschi, B.; Bartsch, H.S.; Sass, S.; Beckers, J.; Theis, F.J.; et al. Quantification of regenerative potential in primary human mammary epithelial cells. Development 2015, 142, 3239–3251. [Google Scholar] [CrossRef] [Green Version]
- Kessel, S.; Cribbes, S.; Dery, O.; Kuksin, D.; Sincoff, E.; Qiu, J.; Chan, L.L. High-Throughput 3D Tumor Spheroid Screening Method for Cancer Drug Discovery Using Celigo Image Cytometry. SLAS Technol. 2017, 22, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Shin, D.; Roh, J.L. Development of an In Vitro cell-sheet cancer model for chemotherapeutic screening. Theranostics 2018, 8, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.; Muller, S.; Stelzer, E.H.; Pampaloni, F. Quantitative 3D cell-based assay performed with cellular spheroids and fluorescence microscopy. Methods Cell Biol. 2013, 113, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Leary, E.; Rhee, C.; Wilks, B.; Morgan, J.R. Accurate quantitative wide-field fluorescence microscopy of 3-D spheroids. Biotechniques 2016, 61, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masselink, W.; Reumann, D.; Murawala, P.; Pasierbek, P.; Taniguchi, Y.; Bonnay, F.; Meixner, K.; Knoblich, J.A.; Tanaka, E.M. Broad applicability of a streamlined ethyl cinnamate-based clearing procedure. Development 2019, 146, dev166884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, J.W.; Conchello, J.A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef]
- Török, T.W.P. Rigorous theory for axial resolution in confocal microscopes. Opt. Commun. 1997, 137, 127–135. [Google Scholar] [CrossRef]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905–1907. [Google Scholar] [CrossRef]
- Gustafsson, M.G.; Shao, L.; Carlton, P.M.; Wang, C.J.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Cullen, D.K.; Gordian-Velez, W.J.; Struzyna, L.A.; Jgamadze, D.; Lim, J.; Wofford, K.L.; Browne, K.D.; Chen, H.I. Bundled Three-Dimensional Human Axon Tracts Derived from Brain Organoids. iScience 2019, 21, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human Brain Organoids on a Chip Reveal the Physics of Folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781.e9. [Google Scholar] [CrossRef]
- Krieger, T.G.; Tirier, S.M.; Park, J.; Jechow, K.; Eisemann, T.; Peterziel, H.; Angel, P.; Eils, R.; Conrad, C. Modeling glioblastoma invasion using human brain organoids and single-cell transcriptomics. Neuro. Oncol. 2020, 22, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer′s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Malatesta, M.; Lien, B.V.; Saha, P.; Thombare, S.S.; Hong, S.J.; Pedraza, L.; Koontz, M.; Seo, K.; Horlbeck, M.A.; et al. CRISPRi-based radiation modifier screen identifies long non-coding RNA therapeutic targets in glioma. Genome Biol. 2020, 21, 83. [Google Scholar] [CrossRef]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.J.; Battula, S.; Zachos, N.; Kovbasnjuk, O.; Fawlke-Abel, J.; In, J.; Donowitz, M.; Verkman, A.S. Microfluidics platform for measurement of volume changes in immobilized intestinal enteroids. Biomicrofluidics 2014, 8, 024106. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Okkelman, I.A.; Foley, T.; Papkovsky, D.B.; Dmitriev, R.I. Live cell imaging of mouse intestinal organoids reveals heterogeneity in their oxygenation. Biomaterials 2017, 146, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Stehbens, S.; Pemble, H.; Murrow, L.; Wittmann, T. Imaging intracellular protein dynamics by spinning disk confocal microscopy. Methods Enzymol. 2012, 504, 293–313. [Google Scholar] [CrossRef] [Green Version]
- Graf, R.; Rietdorf, J.; Zimmermann, T. Live cell spinning disk microscopy. Adv. Biochem. Eng. Biotechnol. 2005, 95, 57–75. [Google Scholar] [CrossRef]
- John Oreopoulos, R.B.; Browne, M. Chapter 9—Spinning-disk confocal microscopy: Present technology and future trends. Methods Cell Biol. 2014, 123, 153–175. [Google Scholar] [CrossRef]
- Lukonin, I.; Serra, D.; Challet Meylan, L.; Volkmann, K.; Baaten, J.; Zhao, R.; Meeusen, S.; Colman, K.; Maurer, F.; Stadler, M.B.; et al. Phenotypic landscape of intestinal organoid regeneration. Nature 2020, 586, 275–280. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Bock, N.; Rohl, J. Real-Time and 3D Quantification of Cancer Cell Dynamics: Exploiting a Bioengineered Human Bone Metastatic Microtissue. Methods Mol. Biol. 2019, 2054, 59–77. [Google Scholar] [CrossRef]
- McKinley, K.L.; Stuurman, N.; Royer, L.A.; Schartner, C.; Castillo-Azofeifa, D.; Delling, M.; Klein, O.D.; Vale, R.D. Cellular aspect ratio and cell division mechanics underlie the patterning of cell progeny in diverse mammalian epithelia. eLife 2018, 7, e36739. [Google Scholar] [CrossRef]
- Conchello, J.A.; Lichtman, J.W. Theoretical analysis of a rotating-disk partially confocal scanning microscope. Appl. Opt. 1994, 33, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Gratton, E.; Barry, N.P.; Beretta, S.; Celli, A. Multiphoton fluorescence microscopy. Methods 2001, 25, 103–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, C.J.R. Multiphoton microscopy: A personal historical review, with some future predictions. J. Biomed. Opt. 2020, 25, 11. [Google Scholar] [CrossRef] [PubMed]
- Favreau, P.F.; He, J.; Gil, D.A.; Deming, D.A.; Huisken, J.; Skala, M.C. Label-free redox imaging of patient-derived organoids using selective plane illumination microscopy. Biomed. Opt. Express 2020, 11, 2591–2606. [Google Scholar] [CrossRef]
- Walsh, A.J.; Skala, M.C. Optical metabolic imaging quantifies heterogeneous cell populations. Biomed. Opt. Express 2015, 6, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Sharick, J.T.; Walsh, C.M.; Sprackling, C.M.; Pasch, C.A.; Pham, D.L.; Esbona, K.; Choudhary, A.; Garcia-Valera, R.; Burkard, M.E.; McGregor, S.M.; et al. Metabolic Heterogeneity in Patient Tumor-Derived Organoids by Primary Site and Drug Treatment. Front. Oncol. 2020, 10, 553. [Google Scholar] [CrossRef]
- Browne, A.W.; Arnesano, C.; Harutyunyan, N.; Khuu, T.; Martinez, J.C.; Pollack, H.A.; Koos, D.S.; Lee, T.C.; Fraser, S.E.; Moats, R.A.; et al. Structural and Functional Characterization of Human Stem-Cell-Derived Retinal Organoids by Live Imaging. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3311–3318. [Google Scholar] [CrossRef]
- Jain, R.K.; Munn, L.L.; Fukumura, D. Dissecting tumour pathophysiology using intravital microscopy. Nat. Rev. Cancer 2002, 2, 266–276. [Google Scholar] [CrossRef]
- Bonapace, L.; Wyckoff, J.; Oertner, T.; Van Rheenen, J.; Junt, T.; Bentires-Alj, M. If you don’t look, you won’t see: Intravital multiphoton imaging of primary and metastatic breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 125–129. [Google Scholar] [CrossRef]
- Rakotoson, I.; Delhomme, B.; Djian, P.; Deeg, A.; Brunstein, M.; Seebacher, C.; Uhl, R.; Ricard, C.; Oheim, M. Fast 3-D Imaging of Brain Organoids With a New Single-Objective Planar-Illumination Two-Photon Microscope. Front. Neuroanat. 2019, 13, 77. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.; Pratiwi, F.W.; Wu, F.C.M.; Chen, P.; Chen, B.C. Recent Progress in Light Sheet Microscopy for Biological Applications. Appl. Spectrosc. 2018, 72, 1137–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaud, E.G.; Peychl, J.; Huisken, J.; Tomancak, P. Guide to light-sheet microscopy for adventurous biologists. Nat. Methods 2015, 12, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J. Optical sectioning microscopy with planar or structured illumination. Nat. Methods 2011, 8, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Planchon, T.A.; Gao, L.; Milkie, D.E.; Davidson, M.W.; Galbraith, J.A.; Galbraith, C.G.; Betzig, E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 2011, 8, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Ji, N. Adaptive optical fluorescence microscopy. Nat. Methods 2017, 14, 374–380. [Google Scholar] [CrossRef]
- Liu, T.L.; Upadhyayula, S.; Milkie, D.E.; Singh, V.; Wang, K.; Swinburne, I.A.; Mosaliganti, K.R.; Collins, Z.M.; Hiscock, T.W.; Shea, J.; et al. Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 2018, 360, eaaq1392. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Dong, X.; Lu, J.; Hu, T.; Pei, G. Constitutive activity of a G protein-coupled receptor, DRD1, contributes to human cerebral organoid formation. Stem Cells 2020, 38, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, H.; Ozaki, Y.; Ashida, T.; Matsubara, T.; Oishi, N.; Kihara, S.; Takahashi, J. Self-Organized Synchronous Calcium Transients in a Cultured Human Neural Network Derived from Cerebral Organoids. Stem Cell Rep. 2019, 13, 458–473. [Google Scholar] [CrossRef] [Green Version]
- Dobosz, M.; Ntziachristos, V.; Scheuer, W.; Strobel, S. Multispectral fluorescence ultramicroscopy: Three-dimensional visualization and automatic quantification of tumor morphology, drug penetration, and antiangiogenic treatment response. Neoplasia 2014, 16, 1–13, W1–W7. [Google Scholar] [CrossRef] [Green Version]
- Hof, L.; Moreth, T.; Koch, M.; Liebisch, T.; Kurtz, M.; Tarnick, J.; Lissek, S.M.; Verstegen, M.M.A.; van der Laan, L.J.W.; Huch, M.; et al. Long-term live imaging and multiscale analysis identify heterogeneity and core principles of epithelial organoid morphogenesis. BMC Biol. 2021, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Für Mikrosk. Anat. 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, H.; Widengren, J. Stimulated Emission Depletion Microscopy. Chem. Rev. 2017, 117, 7377–7427. [Google Scholar] [CrossRef]
- Sloan, S.A.; Andersen, J.; Pasca, A.M.; Birey, F.; Pasca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Hu, J.; Sheng, R.; Lin, Q.; He, X.; Guo, M. Volumetric Compression Induces Intracellular Crowding to Control Intestinal Organoid Growth via Wnt/beta-Catenin Signaling. Cell Stem Cell 2021, 28, 63–78.e7. [Google Scholar] [CrossRef]
- McEvoy, E.; Han, Y.L.; Guo, M.; Shenoy, V.B. Gap junctions amplify spatial variations in cell volume in proliferating tumor spheroids. Nat. Commun. 2020, 11, 6148. [Google Scholar] [CrossRef]
- Albanese, A.; Swaney, J.M.; Yun, D.H.; Evans, N.B.; Antonucci, J.M.; Velasco, S.; Sohn, C.H.; Arlotta, P.; Gehrke, L.; Chung, K. Multiscale 3D phenotyping of human cerebral organoids. Sci. Rep. 2020, 10, 21487. [Google Scholar] [CrossRef]
- Wilson, E.; Knudson, W.; Newell-Litwa, K. Hyaluronan regulates synapse formation and function in developing neural networks. Sci. Rep. 2020, 10, 16459. [Google Scholar] [CrossRef]
- Fang, H.; Geng, S.; Hao, M.; Chen, Q.; Liu, M.; Liu, C.; Tian, Z.; Wang, C.; Takebe, T.; Guan, J.L.; et al. Simultaneous Zn(2+) tracking in multiple organelles using super-resolution morphology-correlated organelle identification in living cells. Nat. Commun. 2021, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.J.A.; Mostaco-Guidolin, L.B. Optical Microscopy and the Extracellular Matrix Structure: A Review. Cells 2021, 10, 1760. [Google Scholar] [CrossRef] [PubMed]
- Moneron, G.; Hell, S.W. Two-photon excitation STED microscopy. Opt. Express 2009, 17, 14567–14573. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Gan, Q.; Ermolayev, V.; Harms, G.S. STED-SPIM: Stimulated emission depletion improves sheet illumination microscopy resolution. Biophys. J. 2011, 100, L43–L45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottanelli, F.; Kromann, E.B.; Allgeyer, E.S.; Erdmann, R.S.; Wood Baguley, S.; Sirinakis, G.; Schepartz, A.; Baddeley, D.; Toomre, D.K.; Rothman, J.E.; et al. Two-colour live-cell nanoscale imaging of intracellular targets. Nat. Commun. 2016, 7, 10778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Michael Salinas, N.F.; Wang, X.; Fan, Y.-Y.; Callaway, E.; Landrock, K.; McMurray, D.; Chapkin, R. High-Fat Diet-Induced Obesity Modulates Colonic Lgr5 + Stem Cell Homeostasis by Dysregulating Plasma Membrane Organization. Curr. Dev. Nutr. 2020, 4, 1684. [Google Scholar] [CrossRef]
- Su, Q.P.; Ju, L.A. Biophysical nanotools for single-molecule dynamics. Biophys. Rev. 2018, 10, 1349–1357. [Google Scholar] [CrossRef]
- Bon, P.; Linares-Loyez, J.; Feyeux, M.; Alessandri, K.; Lounis, B.; Nassoy, P.; Cognet, L. Self-interference 3D super-resolution microscopy for deep tissue investigations. Nat. Methods 2018, 15, 449–454. [Google Scholar] [CrossRef]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzmann, R.; Jovin, T.M.; Cremer, C. Saturated patterned excitation microscopy--a concept for optical resolution improvement. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2002, 19, 1599–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Yao, S.; Chen, Q.; Liu, C.; Cai, Y.; Geng, S.; Bai, Y.; Tian, Z.; Zacharias, A.L.; Takebe, T.; et al. De Novo-Designed Near-Infrared Nanoaggregates for Super-Resolution Monitoring of Lysosomes in Cells, in Whole Organoids, and in Vivo. ACS Nano 2019, 13, 14426–14436. [Google Scholar] [CrossRef]
- Postema, M.M.; Grega-Larson, N.E.; Neininger, A.C.; Tyska, M.J. IRTKS (BAIAP2L1) Elongates Epithelial Microvilli Using EPS8-Dependent and Independent Mechanisms. Curr. Biol. 2018, 28, 2876–2888.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Shao, L.; Higgins, C.D.; Poulton, J.S.; Peifer, M.; Davidson, M.W.; Wu, X.; Goldstein, B.; Betzig, E. Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell 2012, 151, 1370–1385. [Google Scholar] [CrossRef] [Green Version]
- Garita-Hernandez, M.; Guibbal, L.; Toualbi, L.; Routet, F.; Chaffiol, A.; Winckler, C.; Harinquet, M.; Robert, C.; Fouquet, S.; Bellow, S.; et al. Optogenetic Light Sensors in Human Retinal Organoids. Front. Neurosci. 2018, 12, 789. [Google Scholar] [CrossRef]
- Fallet, C.; Caron, J.; Oddos, S.; Tinevez, J.-Y.; Moisan, L.; Sirat, G.Y.; Braitbart, P.O.; Shorte, S.L. Conical diffraction as a versatile building block to implement new imaging modalities for superresolution in fluorescence microscopy. Proceedings of Nanoimaging and Nanospectroscopy II, San Diego, CA, USA, 17–21 August 2014; p. 916905. [Google Scholar]
- Caron, J.; Fallet, C.; Tinevez, J.Y.; Moisan, L.; Braitbart, L.P.; Sirat, G.Y.; Shorte, S.L. Conical diffraction illumination opens the way for low phototoxicity super-resolution imaging. Cell Adh. Migr. 2014, 8, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, F.; Lu, H.; Fang, G.; Wen, S.; Chen, C.; Shan, X.; Xu, X.; Zhang, L.; Stenzel, M.; et al. Super-Resolution Mapping of Single Nanoparticles inside Tumor Spheroids. Small 2020, 16, e1905572. [Google Scholar] [CrossRef]
- Boppart, S.A.; Bouma, B.E.; Pitris, C.; Southern, J.F.; Brezinski, M.E.; Fujimoto, J.G. In Vivo cellular optical coherence tomography imaging. Nat. Med. 1998, 4, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Marschall, S.; Sander, B.; Mogensen, M.; Jorgensen, T.M.; Andersen, P.E. Optical coherence tomography-current technology and applications in clinical and biomedical research. Anal. Bioanal. Chem. 2011, 400, 2699–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhetri, R.K.; Phillips, Z.F.; Troester, M.A.; Oldenburg, A.L. Longitudinal study of mammary epithelial and fibroblast co-cultures using optical coherence tomography reveals morphological hallmarks of pre-malignancy. PLoS ONE 2012, 7, e49148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, D.A.; Deming, D.A.; Skala, M.C. Volumetric growth tracking of patient-derived cancer organoids using optical coherence tomography. Biomed. Opt. Express 2021, 12, 3789–3805. [Google Scholar] [CrossRef] [PubMed]
- Deloria, A.J.; Haider, S.; Dietrich, B.; Kunihs, V.; Oberhofer, S.; Knofler, M.; Leitgeb, R.; Liu, M.; Drexler, W.; Haindl, R. Ultra-High-Resolution 3D Optical Coherence Tomography Reveals Inner Structures of Human Placenta-Derived Trophoblast Organoids. IEEE Trans. Biomed. Eng. 2021, 68, 2368–2376. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Sadek, I.; Miyazawa, A.; Tzu-Wei Shen, L.; Makita, S.; Fukuda, S.; Yamashita, T.; Oka, Y.; Mukherjee, P.; Matsusaka, S.; Oshika, T.; et al. Optical coherence tomography-based tissue dynamics imaging for longitudinal and drug response evaluation of tumor spheroids. Biomed. Opt. Express 2020, 11, 6231–6248. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, A.L.; Yu, X.; Gilliss, T.; Alabi, O.; Taylor, R.M., 2nd; Troester, M.A. Inverse-power-law behavior of cellular motility reveals stromal-epithelial cell interactions in 3D co-culture by OCT fluctuation spectroscopy. Optica 2015, 2, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Klein, O.J.; Wang, H.; Evans, C.L. Longitudinal, label-free, quantitative tracking of cell death and viability in a 3D tumor model with OCT. Sci. Rep. 2016, 6, 27017. [Google Scholar] [CrossRef]
- McLelland, B.T.; Lin, B.; Mathur, A.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Transplanted hESC-Derived Retina Organoid Sheets Differentiate, Integrate, and Improve Visual Function in Retinal Degenerate Rats. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2586–2603. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; McLelland, B.T.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Retina Organoid Transplants Develop Photoreceptors and Improve Visual Function in RCS Rats With RPE Dysfunction. Investig. Ophthalmol. Vis. Sci. 2020, 61, 34. [Google Scholar] [CrossRef]
- Seiler, M.J.; Lin, R.E.; McLelland, B.T.; Mathur, A.; Lin, B.; Sigman, J.; De Guzman, A.T.; Kitzes, L.M.; Aramant, R.B.; Thomas, B.B. Vision Recovery and Connectivity by Fetal Retinal Sheet Transplantation in an Immunodeficient Retinal Degenerate Rat Model. Investig. Ophthalmol. Vis. Sci. 2017, 58, 614–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholler, J.; Groux, K.; Goureau, O.; Sahel, J.A.; Fink, M.; Reichman, S.; Boccara, C.; Grieve, K. Dynamic full-field optical coherence tomography: 3D live-imaging of retinal organoids. Light Sci. Appl. 2020, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Yamaguchi, Y.; Kishi, S. Improved visualization of Henle fiber layer by changing the measurement beam angle on optical coherence tomography. Retina 2011, 31, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Walsh, A.C.; Keane, P.A.; Heussen, F.M.; Pappuru, R.K.; Sadda, S.R. Different phenotypes of the appearance of the outer plexiform layer on optical coherence tomography. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.; Vabre, L.; Boccara, A.C.; Beaurepaire, E. High-resolution full-field optical coherence tomography with a Linnik microscope. Appl. Opt. 2002, 41, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.; Grieve, K.; Moneron, G.; Lecaque, R.; Vabre, L.; Boccara, C. Ultrahigh-resolution full-field optical coherence tomography. Appl. Opt. 2004, 43, 2874–2883. [Google Scholar] [CrossRef]
- Mece, P.; Scholler, J.; Groux, K.; Boccara, C. High-resolution In-Vivo human retinal imaging using full-field OCT with optical stabilization of axial motion. Biomed. Opt. Express 2020, 11, 492–504. [Google Scholar] [CrossRef]
- Apelian, C.; Harms, F.; Thouvenin, O.; Boccara, A.C. Dynamic full field optical coherence tomography: Subcellular metabolic contrast revealed in tissues by interferometric signals temporal analysis. Biomed. Opt. Express 2016, 7, 1511–1524. [Google Scholar] [CrossRef] [Green Version]
- Groux, K.; Verschueren, A.; Nanteau, C.; Clémençon, M.; Fink, M.; Sahel, J.-A.; Boccara, C.; Paques, M.; Reichman, S.; Grieve, K. Non invasive live imaging of a novel retinal pigment epithelium stress model with Dynamic Full-Field Optical Coherence Tomography. arXiv 2021, arXiv:2106.10531. [Google Scholar]
- Leroux, C.E.; Bertillot, F.; Thouvenin, O.; Boccara, A.C. Intracellular dynamics measurements with full field optical coherence tomography suggest hindering effect of actomyosin contractility on organelle transport. Biomed. Opt. Express 2016, 7, 4501–4513. [Google Scholar] [CrossRef] [Green Version]
- Scholler, J. Motion artifact removal and signal enhancement to achieve in vivo dynamic full field OCT. Opt. Express 2019, 27, 19562–19572. [Google Scholar] [CrossRef] [PubMed]
- Zernike, F. How I discovered phase contrast. Science 1955, 121, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Lee, S.; Choi, W.H.; Jee, J.H.; Kim, H.R.; Yoo, J. Fabrication of Dentin-Pulp-Like Organoids Using Dental-Pulp Stem Cells. Cells 2020, 9, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003, 361, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Ha, A.; de Lau, W.; Yuki, K.; Santos, A.J.M.; You, C.; Geurts, M.H.; Puschhof, J.; Pleguezuelos-Manzano, C.; Peng, W.C.; et al. Next-Generation Surrogate Wnts Support Organoid Growth and Deconvolute Frizzled Pleiotropy In Vivo. Cell Stem Cell 2020, 27, 840–851.e6. [Google Scholar] [CrossRef]
- Vig, A.; Poulter, J.A.; Ottaviani, D.; Tavares, E.; Toropova, K.; Tracewska, A.M.; Mollica, A.; Kang, J.; Kehelwathugoda, O.; Paton, T.; et al. DYNC2H1 hypomorphic or retina-predominant variants cause nonsyndromic retinal degeneration. Genet. Med. 2020, 22, 2041–2051. [Google Scholar] [CrossRef]
- Akkerman, N.; Defize, L.H. Dawn of the organoid era: 3D tissue and organ cultures revolutionize the study of development, disease, and regeneration. Bioessays 2017, 39, 1600244. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef] [Green Version]
- Pettinato, G.; Coughlan, M.F.; Zhang, X.; Chen, L.; Khan, U.; Glyavina, M.; Sheil, C.J.; Upputuri, P.K.; Zakharov, Y.N.; Vitkin, E.; et al. Spectroscopic label-free microscopy of changes in live cell chromatin and biochemical composition in transplantable organoids. Sci. Adv. 2021, 7, eabj2800. [Google Scholar] [CrossRef]
- Zong, W.; Wu, R.; Li, M.; Hu, Y.; Li, Y.; Li, J.; Rong, H.; Wu, H.; Xu, Y.; Lu, Y.; et al. Fast high-resolution miniature two-photon microscopy for brain imaging in freely behaving mice. Nat. Methods 2017, 14, 713–719. [Google Scholar] [CrossRef]
- Zong, W.; Zhao, J.; Chen, X.; Lin, Y.; Ren, H.; Zhang, Y.; Fan, M.; Zhou, Z.; Cheng, H.; Sun, Y.; et al. Large-field high-resolution two-photon digital scanned light-sheet microscopy. Cell Res. 2015, 25, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Fan, J.; Li, L.; Liu, H.; Wu, R.; Wu, Y.; Wei, L.; Mao, H.; Lal, A.; Xi, P.; et al. Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nat. Biotechnol. 2018, 36, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Linarès-Loyez, J.; Ferreira, J.S.; Rossier, O.; Lounis, B.; Giannone, G.; Groc, L.; Cognet, L.; Bon, P. Self-interference (SELFI) microscopy for live super-resolution imaging and single particle tracking in 3D. Front. Phys. 2019, 7, 68. [Google Scholar] [CrossRef]
- Truong, T.V.; Supatto, W.; Koos, D.S.; Choi, J.M.; Fraser, S.E. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat. Methods 2011, 8, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Mahou, P.; Vermot, J.; Beaurepaire, E.; Supatto, W. Multicolor two-photon light-sheet microscopy. Nat. Methods 2014, 11, 600–601. [Google Scholar] [CrossRef]
- Fahrbach, F.O.; Gurchenkov, V.; Alessandri, K.; Nassoy, P.; Rohrbach, A. Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation. Opt. Express 2013, 21, 13824–13839. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Kim, D.; Kwon, H.S. Oblique scanning 2-photon light-sheet fluorescence microscopy for rapid volumetric imaging. J. Biophotonics 2018, 11, e201700270. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Liu, Y.-L.; Bi, S.; Huang, S.-P.; Borrego, E.D.; Chen, Y.-I.; Kuo, Y.-A.; Yeh, H.-C. Single-objective multiphoton light-sheet microscopy for tumor organoid screening. In Proceedings of the Multiphoton Microscopy in the Biomedical Sciences XIX, San Francisco, CA, USA, 2–7 February 2019; p. 108822P. [Google Scholar]
- van de Linde, S.; Heilemann, M.; Sauer, M. Live-cell super-resolution imaging with synthetic fluorophores. Annu. Rev. Phys. Chem. 2012, 63, 519–540. [Google Scholar] [CrossRef]
- Lukinavicius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Correa, I.R., Jr.; Luo, Z.G.; et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef]
- Lukinavicius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Gottfert, F.; Ta, H.; Guther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef]
- Grimm, J.B.; English, B.P.; Choi, H.; Muthusamy, A.K.; Mehl, B.P.; Dong, P.; Brown, T.A.; Lippincott-Schwartz, J.; Liu, Z.; Lionnet, T.; et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 2016, 13, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Itzkan, I.; Qiu, L.; Fang, H.; Zaman, M.M.; Vitkin, E.; Ghiran, I.C.; Salahuddin, S.; Modell, M.; Andersson, C.; Kimerer, L.M.; et al. Confocal light absorption and scattering spectroscopic microscopy monitors organelles in live cells with no exogenous labels. Proc. Natl. Acad. Sci. USA 2007, 104, 17255–17260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleskow, D.K.; Zhang, L.; Turzhitsky, V.; Coughlan, M.F.; Khan, U.; Zhang, X.; Sheil, C.J.; Glyavina, M.; Chen, L.; Shinagare, S.; et al. Coherent confocal light scattering spectroscopic microscopy evaluates cancer progression and aggressiveness in live cells and tissue. ACS Photonics 2021, 8, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Coutu, D.L.; Kokkaliaris, K.D.; Kunz, L.; Schroeder, T. Multicolor quantitative confocal imaging cytometry. Nat. Methods 2018, 15, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Germain, R.N.; Gerner, M.Y. High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat. Protoc. 2019, 14, 1708–1733. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, J.C.; Cooper, S.; Heigwer, F.; Warchal, S.; Qiu, P.; Molnar, C.; Vasilevich, A.S.; Barry, J.D.; Bansal, H.S.; Kraus, O.; et al. Data-analysis strategies for image-based cell profiling. Nat. Methods 2017, 14, 849–863. [Google Scholar] [CrossRef]
- Bian, X.; Li, G.; Wang, C.; Liu, W.; Lin, X.; Chen, Z.; Cheung, M.; Luo, X. A deep learning model for detection and tracking in high-throughput images of organoid. Comput. Biol. Med. 2021, 134, 104490. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Resolution | Penetration Depth | 3D | Living Cell Imaging | Photobleaching/Phototoxicity | Advantage | Disadvantage | Application |
|---|---|---|---|---|---|---|---|---|
| EM | ~0.1 nm | ~150 nm | ± | − | − | Nanometer resolution | Damage sample | Precise observation of ultrastructure |
| Bright-field microscopy | ~2 µm | − | − | + | − |
| 3D information is not captured |
|
| WFFM | 200–300 nm (XY) 500–700 nm (Z) | Bad | − | + | Low |
|
|
|
| LSCM | ≈200 nm (XY) 350–800 nm (Z) | ≈100 µm | + | + | High |
|
|
|
| SDC microscopy | <LSCM | >LSCM | + | + | Lower than LSCM |
|
|
|
| Multiphoton microscopy | ≈LSCM | Hundreds of µm | + | + | Low, restricted to focal plane |
|
|
|
| LSFM | <LSCM | >Multiphoton microscopy | + | + | Low, restricted to focal plane |
|
|
|
| STED | 50 nm (XY) 100 nm (Z) | <50 μm | + | Not suitable | High |
| High laser intensity leads to serious photobleaching and phototoxicity |
|
| SMLM | 20 nm (XY) 50 nm (Z) | <a few of μm | − | − | Serious than STED |
|
|
|
| SIM | 100–150 nm (XY) ~300 nm (Z) | <50 μm | + | + | Low |
|
|
|
| OCT | 10 µm | 1–3 mm | + | + | − |
|
|
|
| FFOCT | 0.5 µm (XY) 1 µm (Z) | 1 mm | + | + | − |
| Lack of functional information |
|
| D-FFOCT | 0.5 µm (XY) 1 µm (Z) | 100 µm | + | + | − |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fei, K.; Zhang, J.; Yuan, J.; Xiao, P. Present Application and Perspectives of Organoid Imaging Technology. Bioengineering 2022, 9, 121. https://doi.org/10.3390/bioengineering9030121
Fei K, Zhang J, Yuan J, Xiao P. Present Application and Perspectives of Organoid Imaging Technology. Bioengineering. 2022; 9(3):121. https://doi.org/10.3390/bioengineering9030121
Chicago/Turabian StyleFei, Keyi, Jinze Zhang, Jin Yuan, and Peng Xiao. 2022. "Present Application and Perspectives of Organoid Imaging Technology" Bioengineering 9, no. 3: 121. https://doi.org/10.3390/bioengineering9030121
APA StyleFei, K., Zhang, J., Yuan, J., & Xiao, P. (2022). Present Application and Perspectives of Organoid Imaging Technology. Bioengineering, 9(3), 121. https://doi.org/10.3390/bioengineering9030121