
Systematic Investigation of the Effects of Multiple SV40 Nuclear Localization Signal Fusion on the Genome Editing Activity of Purified SpCas9


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of Cas9 Proteins
2.2. In Vitro Transcription of sgRNA
2.3. In Vitro Cleavage Assay
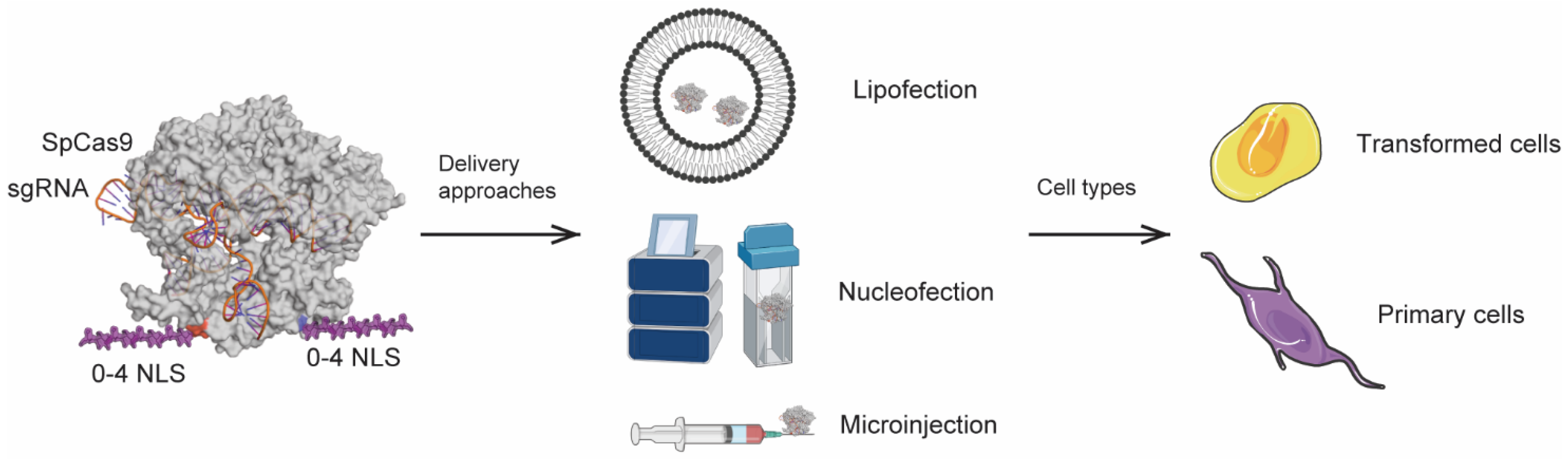
2.4. Cell Lines, Primary Cells and Stem Cells
2.5. Nucleofection
2.6. Lipofection
2.7. Microinjection of Mouse Embryos
2.8. Western Blot
3. Results
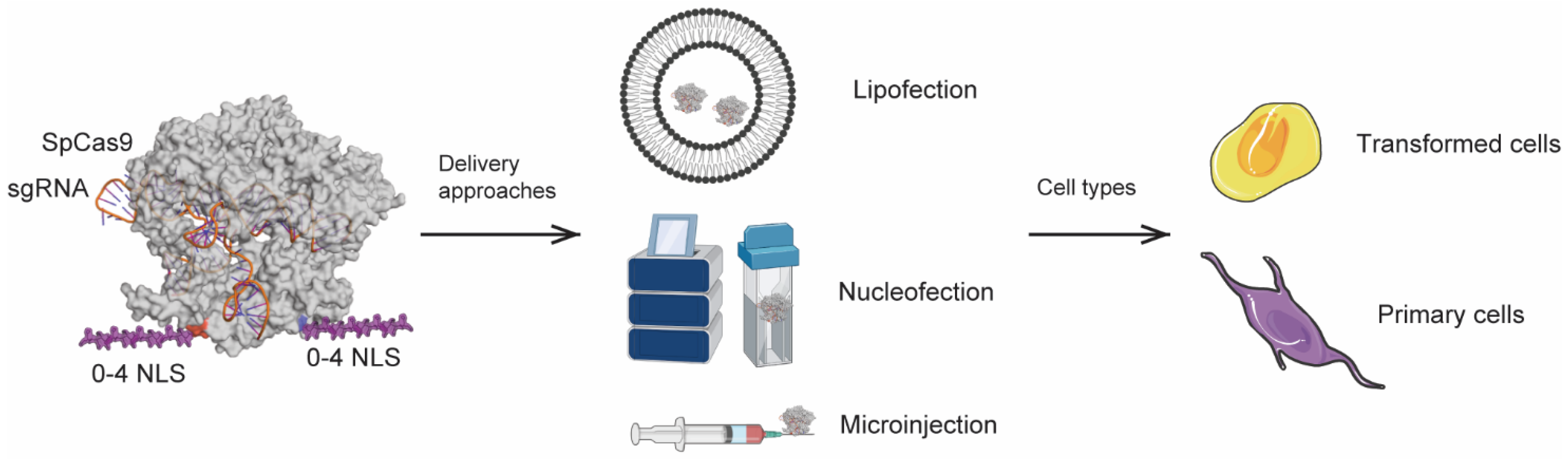
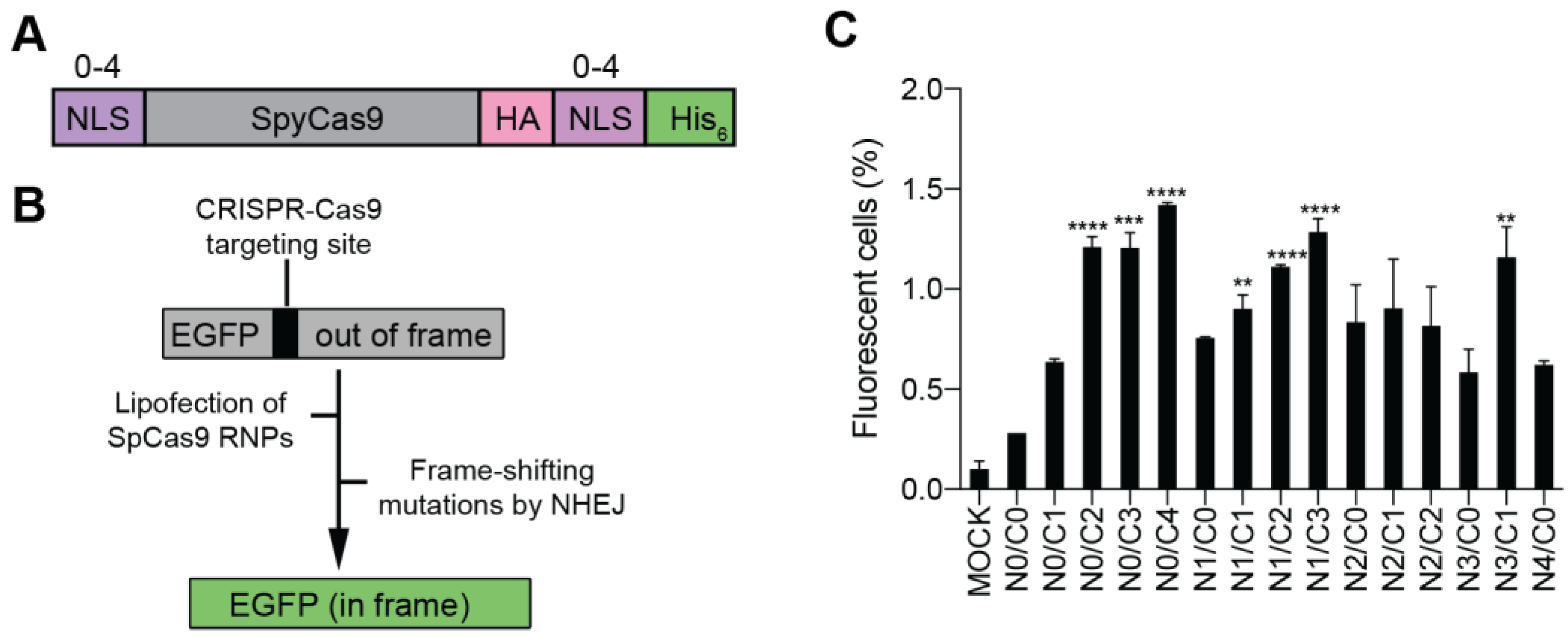
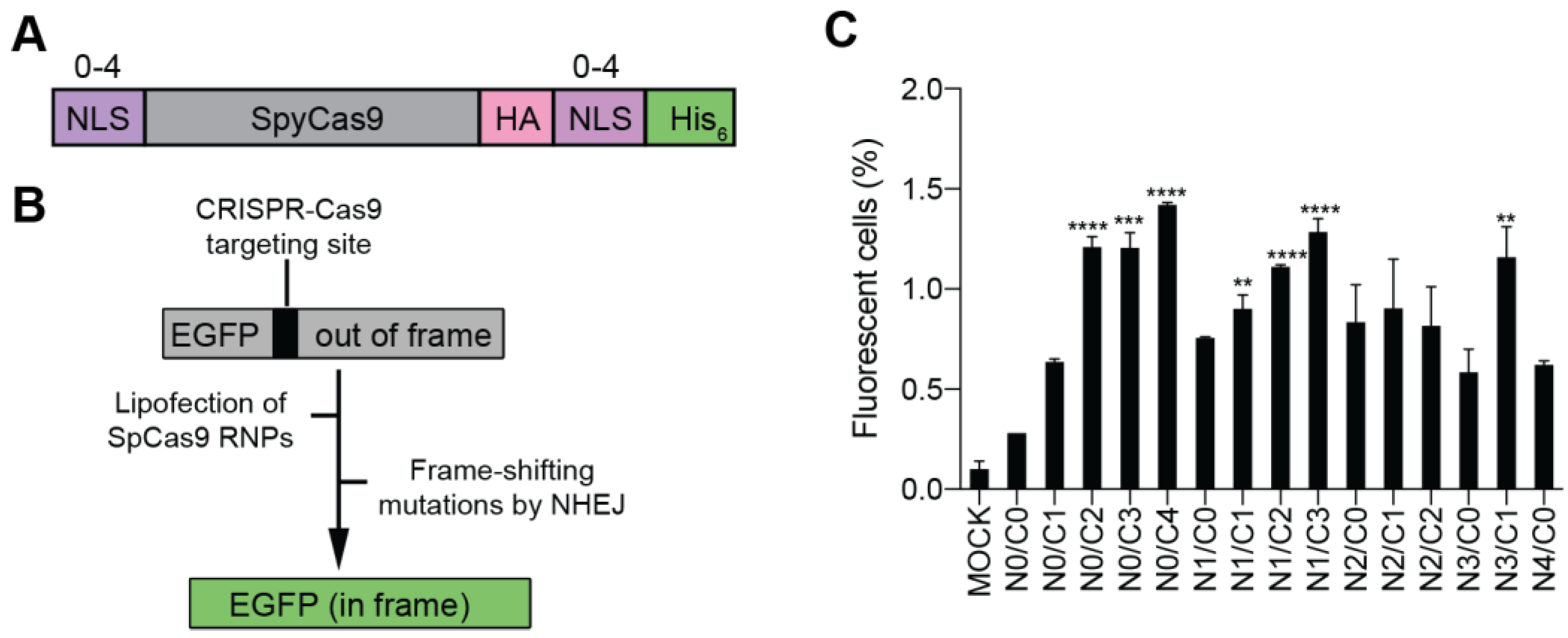
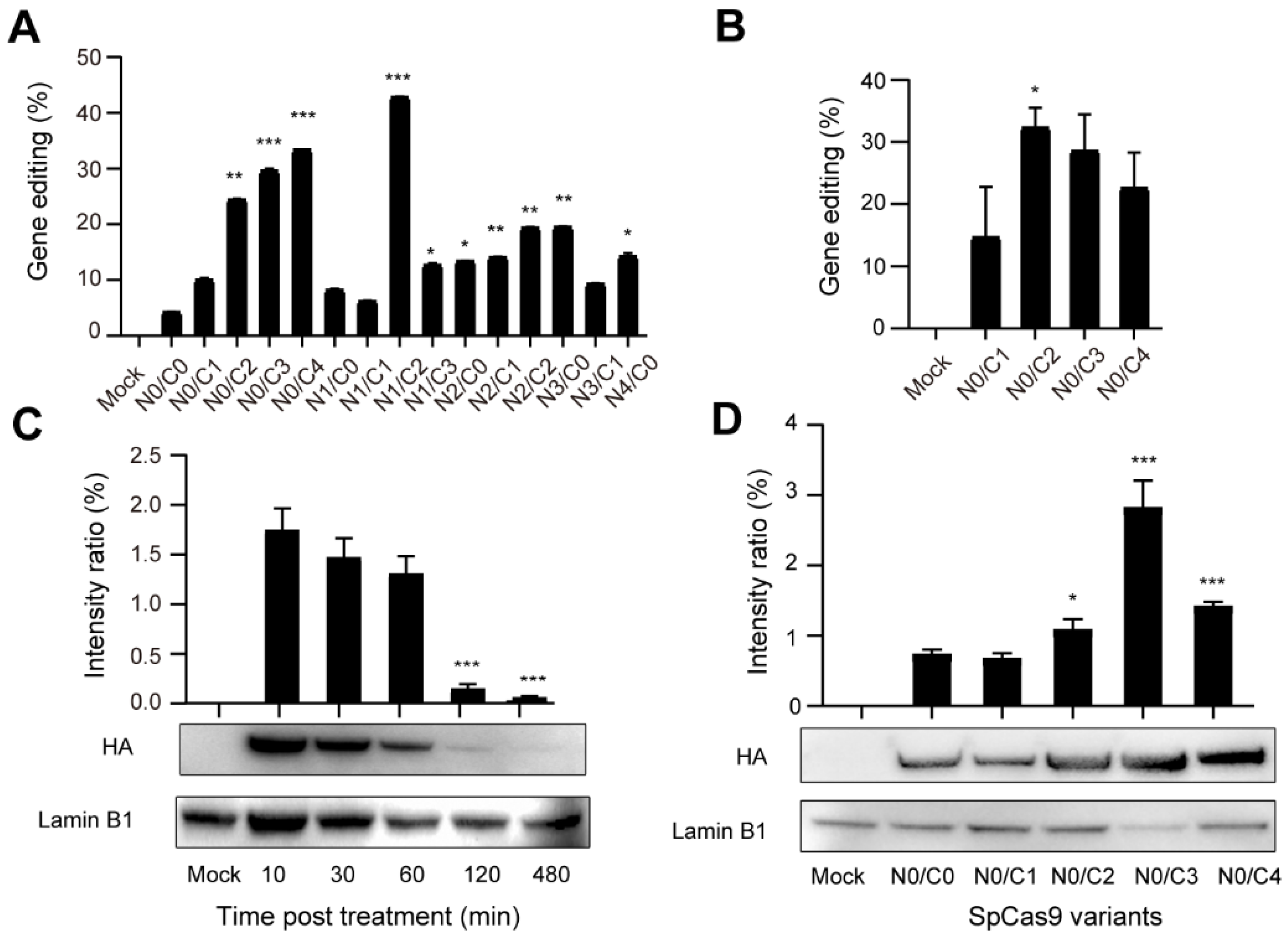
3.1. Production and Validation of Multi-NLS SpCas9 Proteins
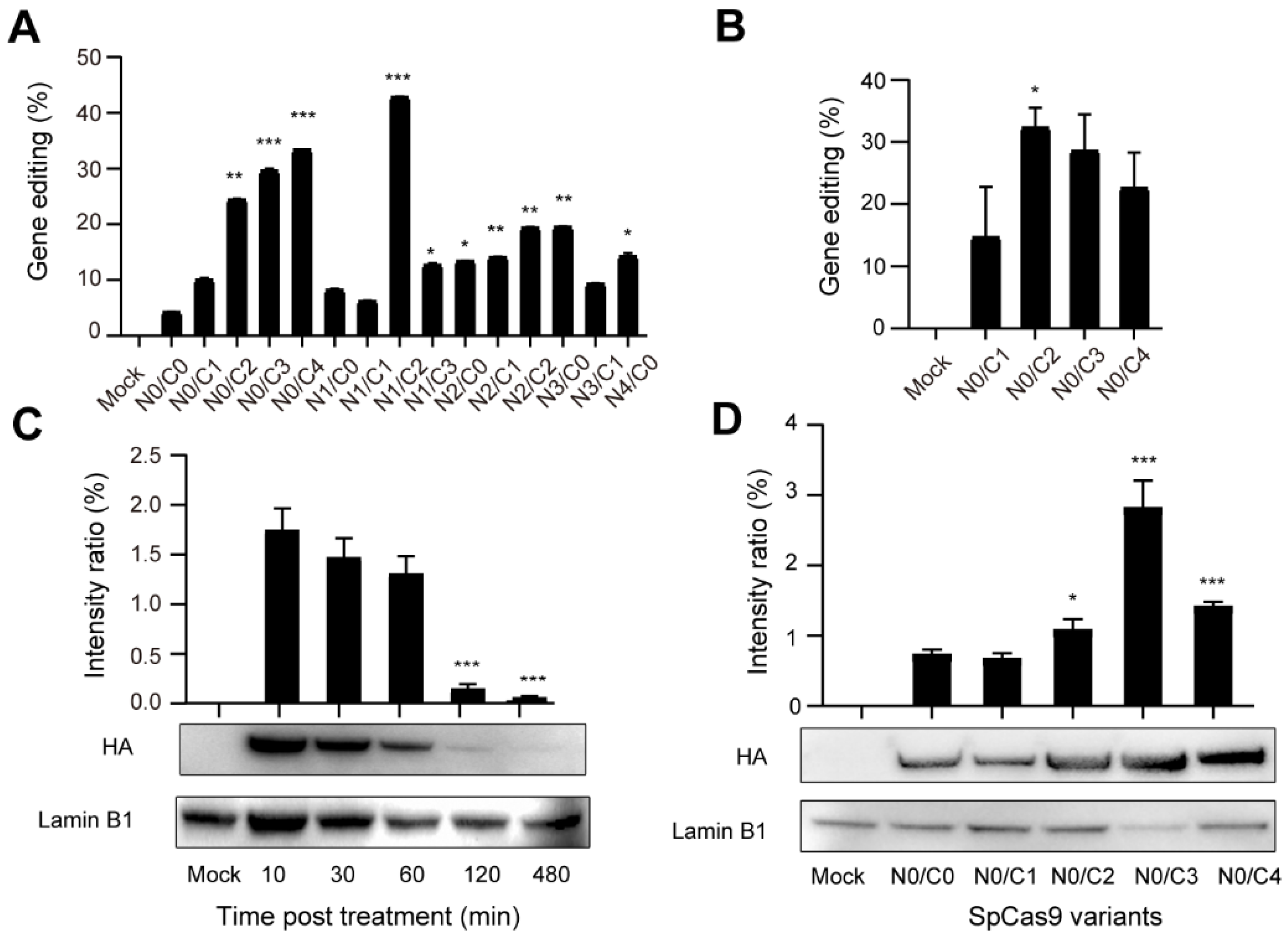
3.2. Evaluation of the Cellular Activity of Lipofected Multi-NLS SpCas9 Proteins in EGFP Reporter Cells
3.3. Evaluation of the Cellular Activity of Nucleofected Multi-NLS SpCas9 Proteins
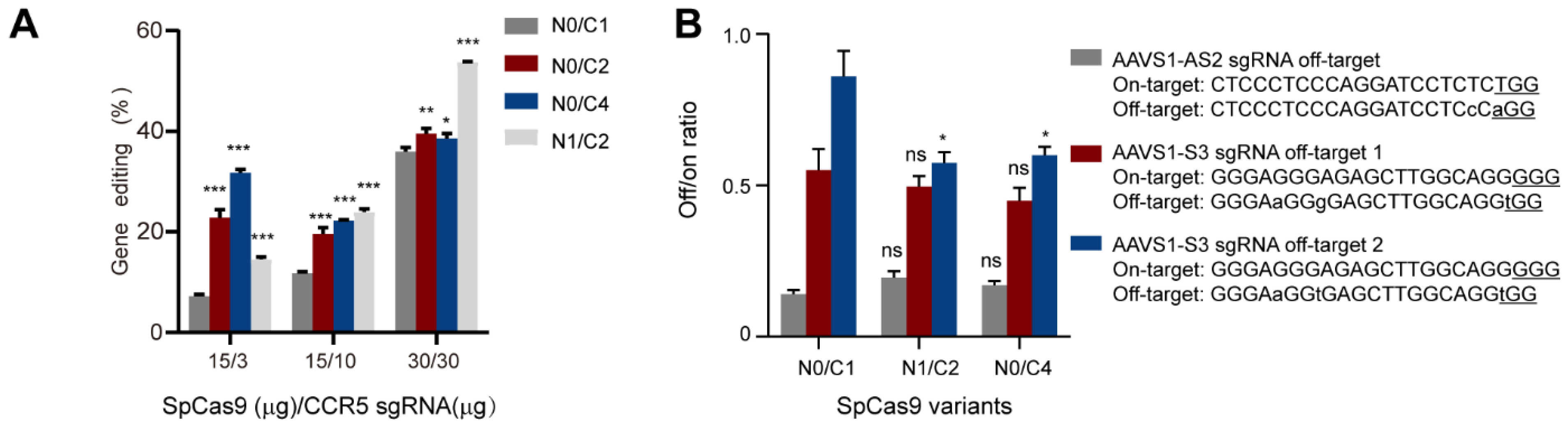
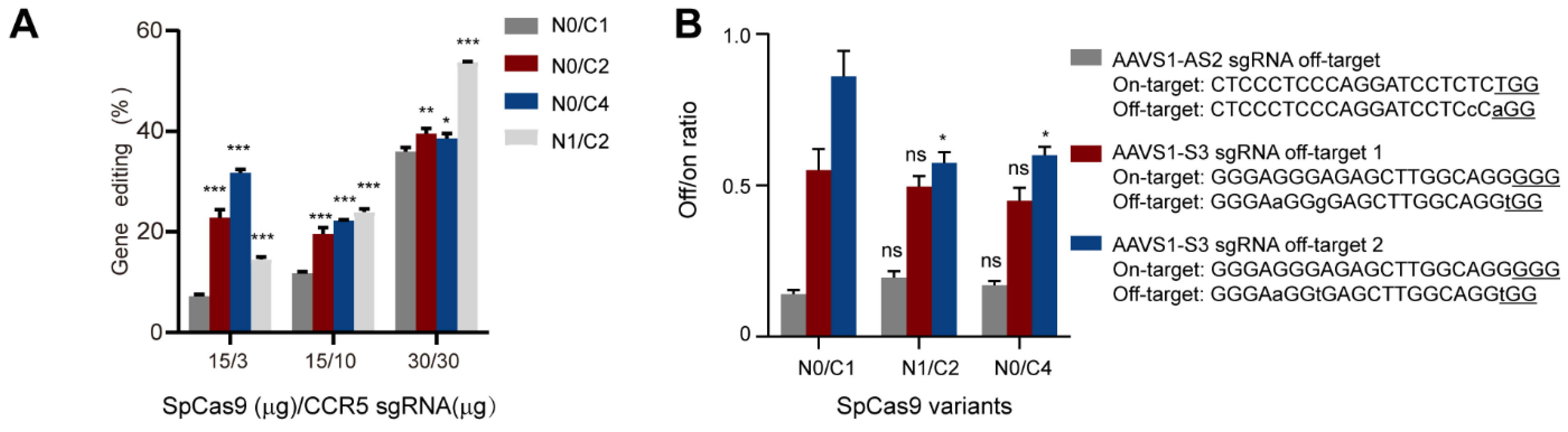
3.4. Evaluation of the Genome-Editing Specificity of Multi-NLS SpCas9 Proteins Delivered via Nucleofection into K562 Cells
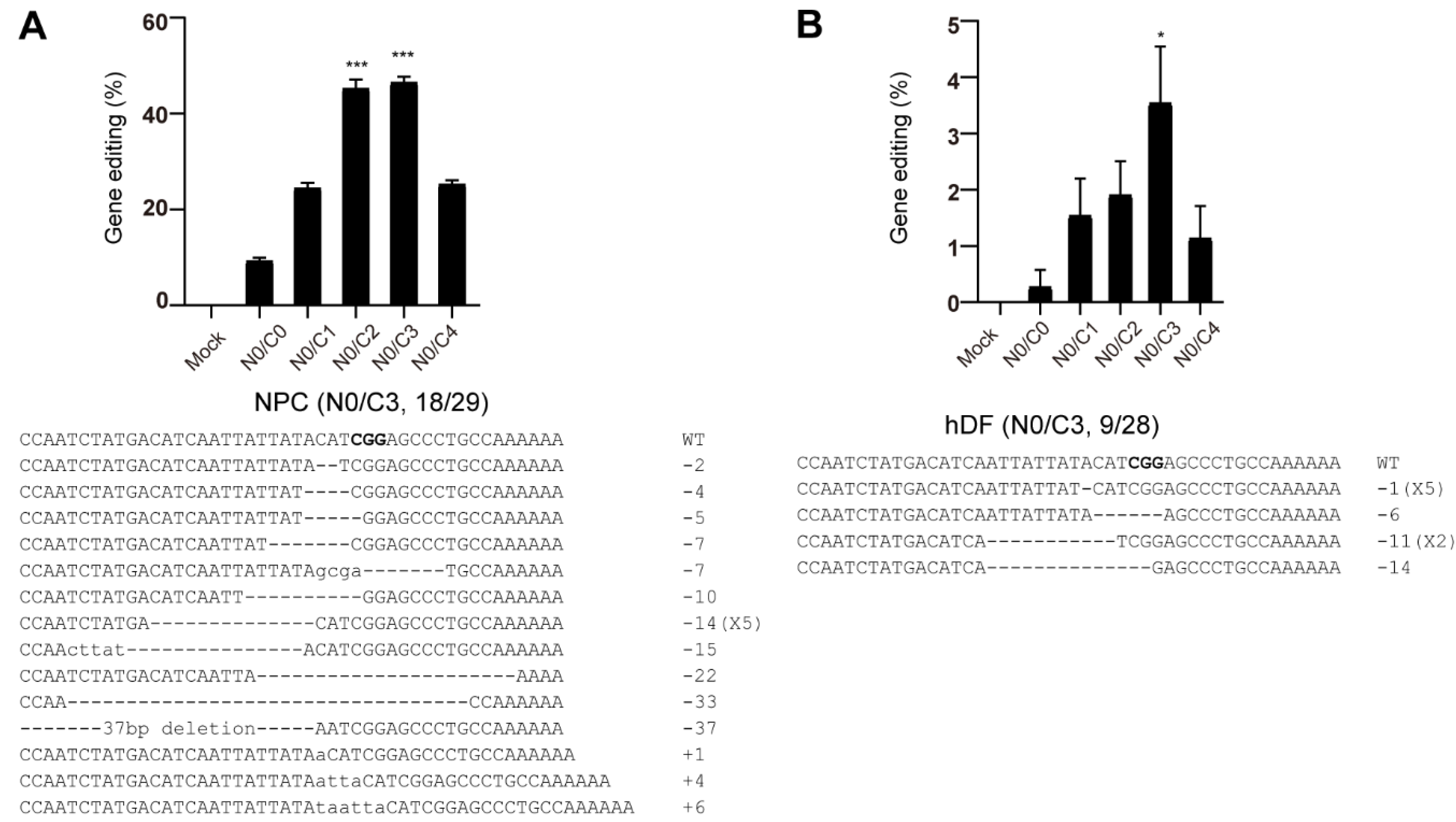
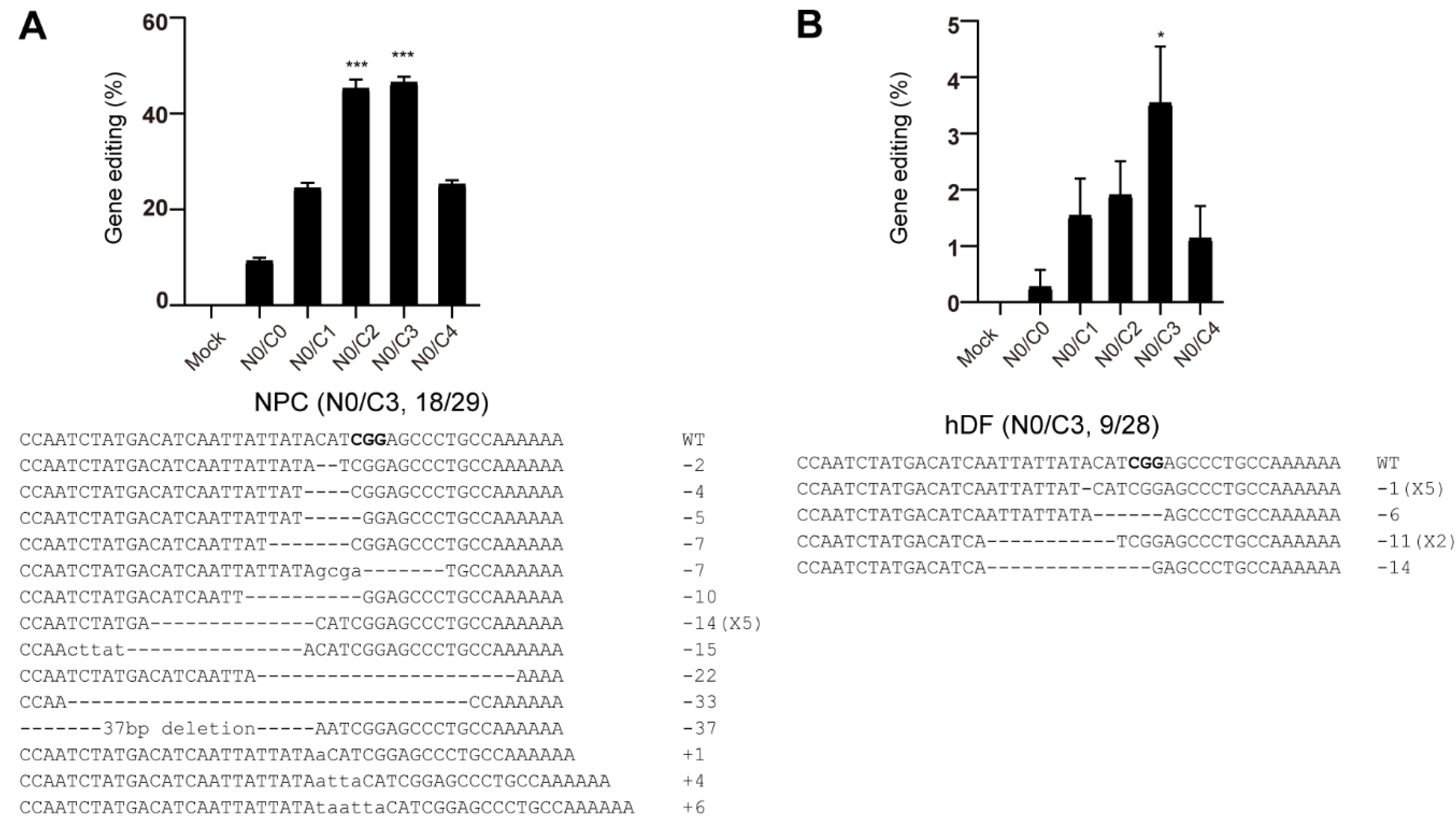
3.5. Genome Editing of Primary Cells Using Nucleofection of Multi-NLS SpCas9 Proteins
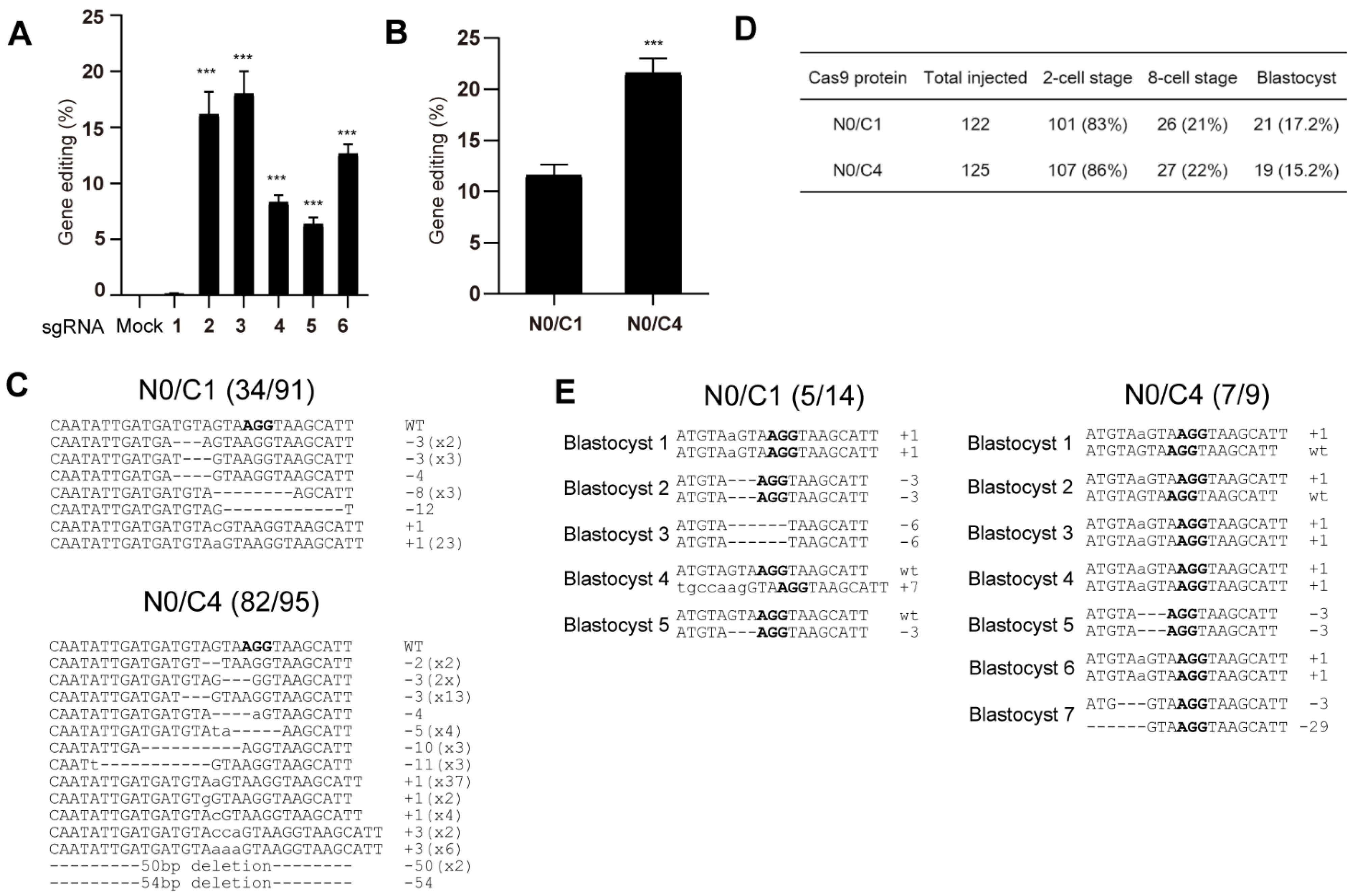
3.6. Genome Editing of Mouse Embryos Using Microinjection of Multi-NLS SpCas9 Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2, e00471. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Garciabustos, J.; Heitman, J.; Hall, M.N. Nuclear protein localization. Biochim. Biophys. Acta 1991, 1071, 83–101. [Google Scholar] [CrossRef]
- Gaj, T.; Sirk, S.J.; Shui, S.L.; Liu, J. Genome-editing technologies: Principles and applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Gaj, T.; Wallen, M.C.; Barbas, C.F., 3rd. Improved cell-penetrating zinc-finger nuclease proteins for precision genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e232. [Google Scholar] [CrossRef]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryo. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Naseri, A.; Reyes-Gutierrez, P.; Wolfe, S.A.; Zhang, S.; Pederson, T. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, 3002–3007. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Cui, H.; Ying, L.; Yu, X.F. Enhanced cytosolic delivery and release of CRISPR/Cas9 by black phosphorus nanosheets for genome editing. Angew. Chem. Int. Ed. 2018, 57, 10268–10272. [Google Scholar] [CrossRef]
- Staahl, B.T.; Benekareddy, M.; Coulon-Bainier, C.; Banfal, A.A.; Floor, S.N.; Sabo, J.K.; Urnes, C.; Munares, G.A.; Ghosh, A.; Doudna, J.A. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017, 35, 431–434. [Google Scholar] [CrossRef]
- Hu, P.N.; Zhao, X.Y.; Zhang, Q.H.; Li, W.M.; Zu, Y. Comparison of various nuclear localization signal-fused Cas9 proteins and Cas9 mRNA for genome editing in Zebrafish. G3-Genes Genomes Genet. 2018, 8, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Luk, K.; Shin, M.; Idrizi, F.; Kwok, S.; Roscoe, B.; Mintzer, E.; Suresh, S.; Morrison, K.; Frazao, J.B.; et al. Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res. 2019, 47, 4169–4180. [Google Scholar] [CrossRef] [Green Version]
- Maggio, I.; Zittersteijn, H.A.; Wang, Q.; Liu, J.; Janssen, J.M.; Ojeda, I.T.; van der Maarel, S.M.; Lankester, A.C.; Hoeben, R.C.; Goncalves, M. Integrating gene delivery and gene-editing technologies by adenoviral vector transfer of optimized CRISPR-Cas9 components. Gene Ther. 2020, 27, 209–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Gaj, T.; Yang, Y.; Wang, N.; Shui, S.; Kim, S.; Kanchiswamy, C.N.; Kim, J.S.; Barbas, C.F., 3rd. Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat. Protoc. 2015, 10, 1842–1859. [Google Scholar] [CrossRef] [PubMed]
- Wefers, B.; Panda, S.K.; Ortiz, O.; Brandl, C.; Hensler, S.; Hansen, J.; Wurst, W.; Kuhn, R. Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat. Protoc. 2013, 8, 2355–2379. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shui, S.; Wang, S.; Liu, J. Systematic Investigation of the Effects of Multiple SV40 Nuclear Localization Signal Fusion on the Genome Editing Activity of Purified SpCas9. Bioengineering 2022, 9, 83. https://doi.org/10.3390/bioengineering9020083
Shui S, Wang S, Liu J. Systematic Investigation of the Effects of Multiple SV40 Nuclear Localization Signal Fusion on the Genome Editing Activity of Purified SpCas9. Bioengineering. 2022; 9(2):83. https://doi.org/10.3390/bioengineering9020083
Chicago/Turabian StyleShui, Sailan, Shaojie Wang, and Jia Liu. 2022. "Systematic Investigation of the Effects of Multiple SV40 Nuclear Localization Signal Fusion on the Genome Editing Activity of Purified SpCas9" Bioengineering 9, no. 2: 83. https://doi.org/10.3390/bioengineering9020083
APA StyleShui, S., Wang, S., & Liu, J. (2022). Systematic Investigation of the Effects of Multiple SV40 Nuclear Localization Signal Fusion on the Genome Editing Activity of Purified SpCas9. Bioengineering, 9(2), 83. https://doi.org/10.3390/bioengineering9020083