
Design and Evaluation of Engineered Extracellular Vesicle (EV)-Based Targeting for EGFR-Overexpressing Tumor Cells Using Monobody Display

 , , and
, , and
Abstract
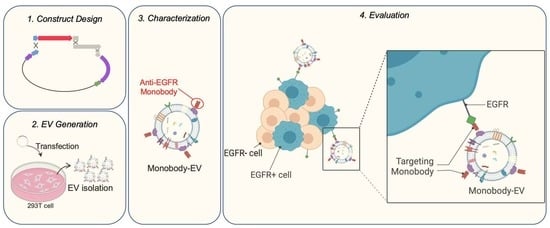
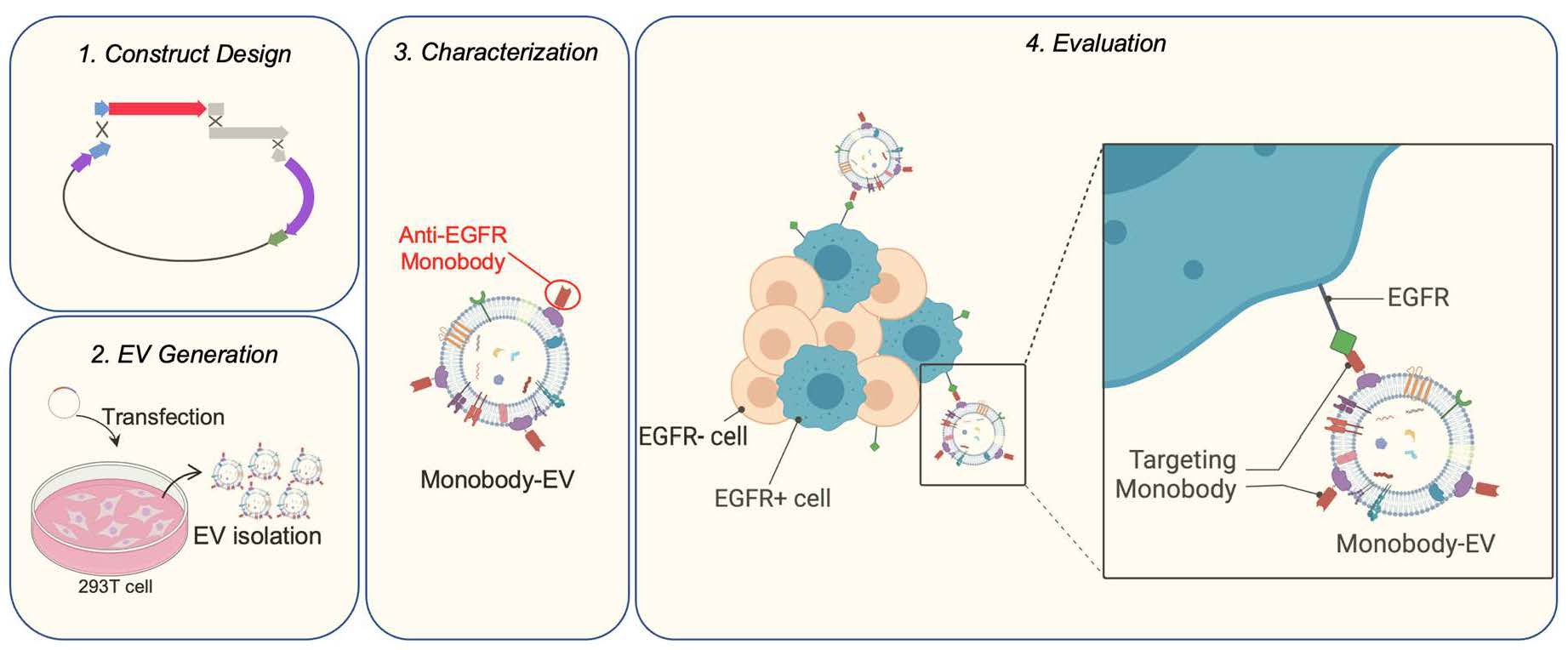{kind=link}
{kind=link}
{kind=link}
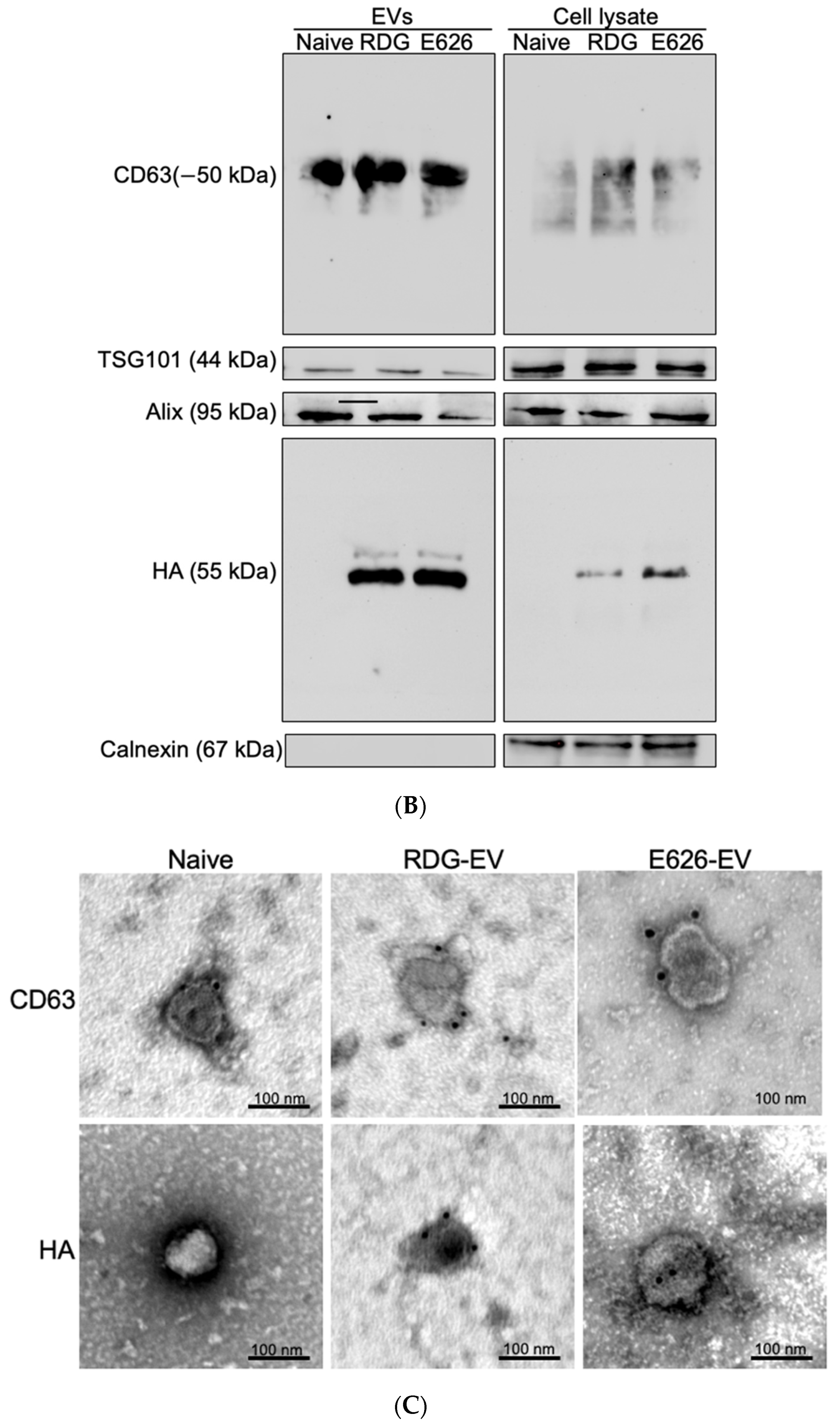{kind=link}
{kind=link}
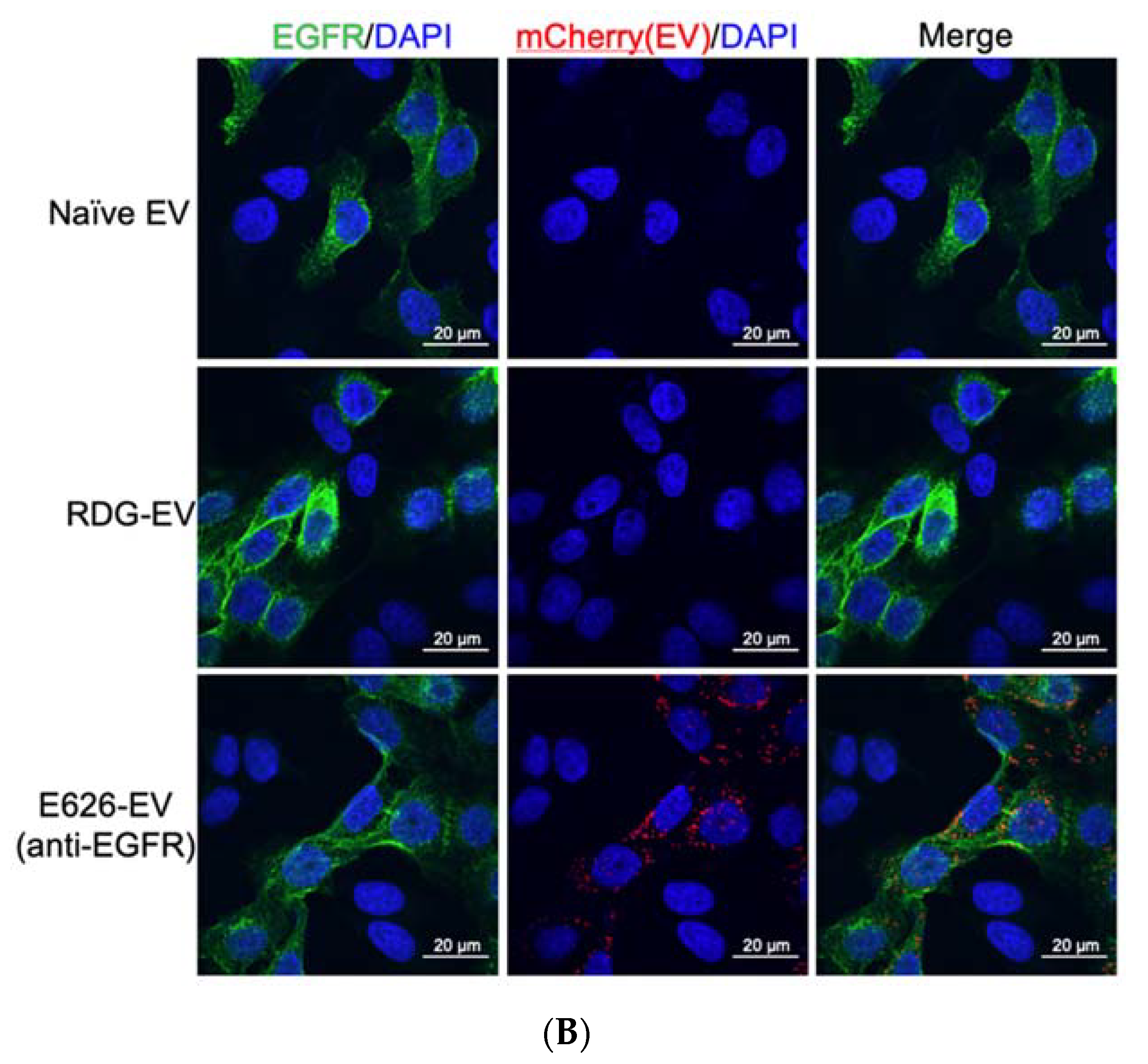{kind=link}
1. Introduction
2. Materials and Methods
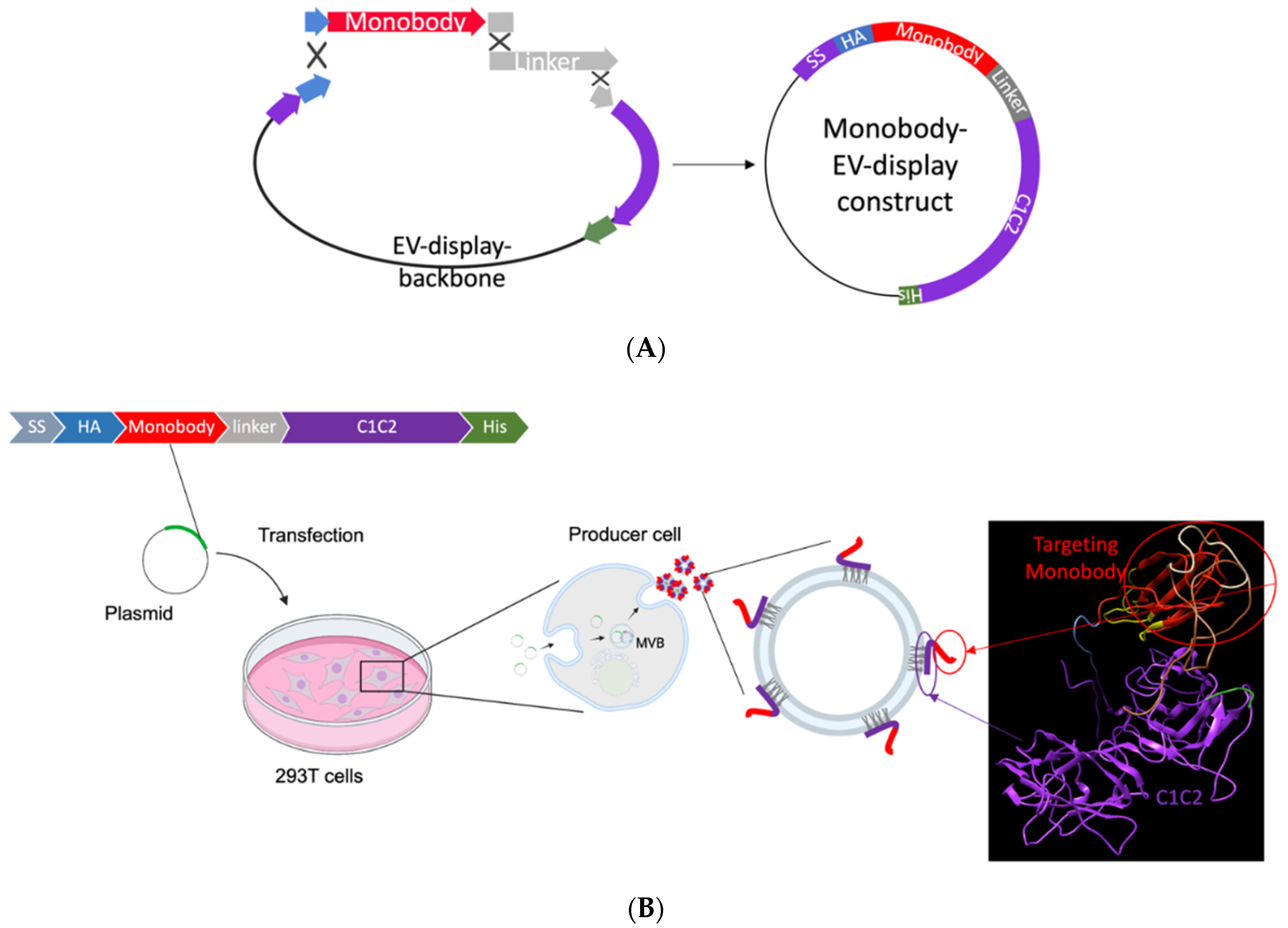
2.1. EV Monobody Display Plasmid Construction
2.2. Cell Culture and Treatment
2.3. EV Isolation
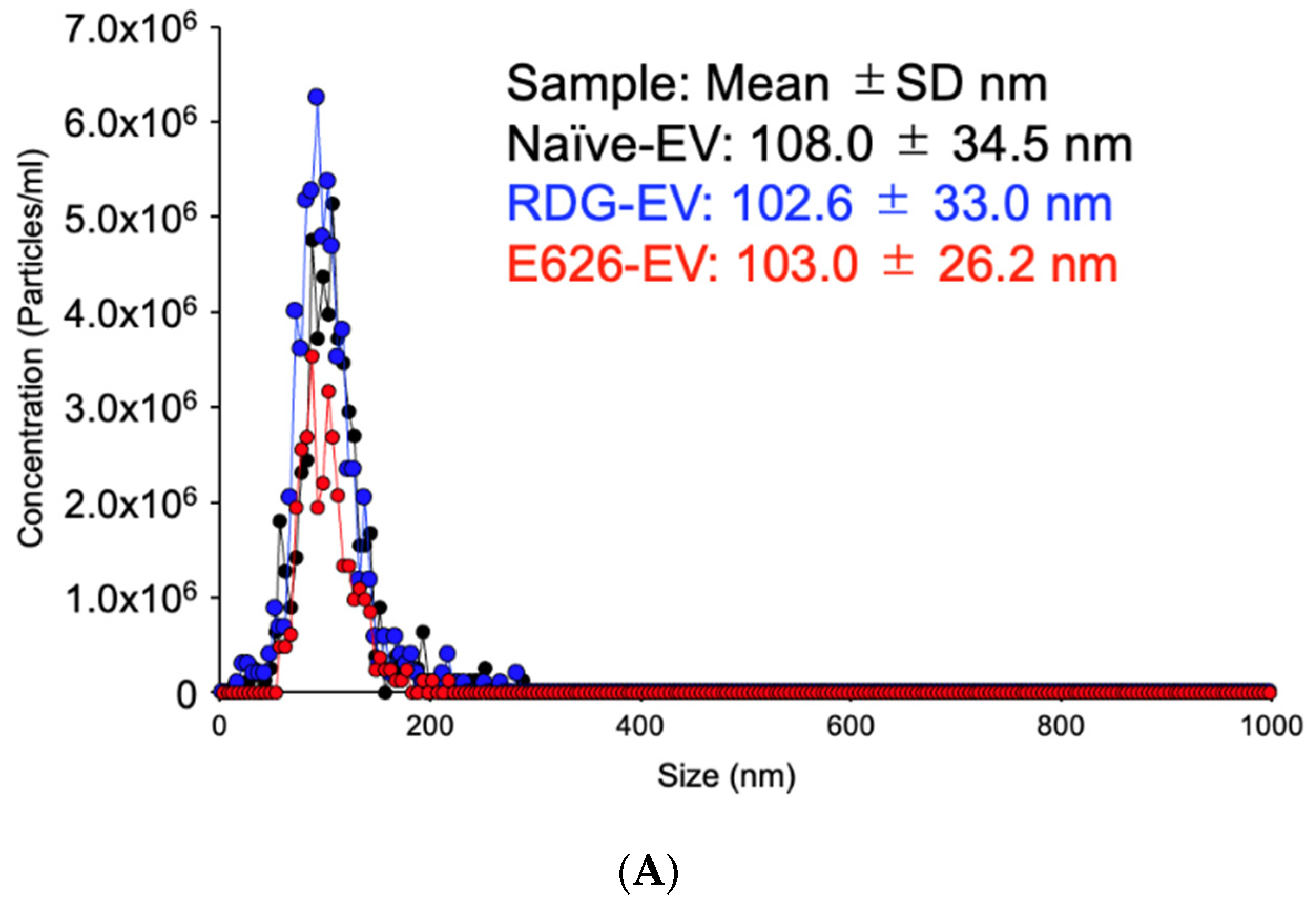
2.4. Nanoparticle Tracking Analysis (NTA)
2.5. Western Blotting
2.6. Immuno-Transmission Electron Microscopy (Immuno-TEM)
2.7. Bioluminescence Assay
2.8. Confocal Microscopy
3. Results
3.1. EV Surface Engineering Strategy and Design
3.2. EV Generation and Characterization
3.3. Engineering EVs for Imaging and Visualization
3.4. In Vitro Evaluation of EV Binding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EVs | extracellular vesicles |
| EGFR | epidermal growth factor receptor |
| HER2 | human epidermal growth factor receptor 2 |
| MFG-E8 | milk fat globule EGF factor VIII |
| CDR | complementarity-determining regions |
| MVs | microvesicles |
| PS | phosphatidylserine |
| MVB | multivesicular body |
| SLiCE | Seamless Ligation Cloning Extract |
| FN3 | fibronectin type III |
| gLuc | gaussia luciferase |
| IVIS | in vivo imaging system |
| NTA | nanoparticle tracking analysis |
| Immuno-TEM | immuno-transmission electron microscopy |
References
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Krumeich, S.; Raposo, G.; Théry, C. Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J. Extracell. Vesicles 2012, 1, 1. [Google Scholar] [CrossRef]
- Mall, C.; Rocke, D.M.; Durbin-Johnson, B.; Weiss, R.H. Stability of miRNA in human urine supports its biomarker potential. Biomarkers Med. 2013, 7, 623–631. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Kosaka, N.; Izumi, H.; Sekine, K.; Ochiya, T. microRNA as a new immune-regulatory agent in breast milk. Silence 2010, 1, 7–8. [Google Scholar] [CrossRef]
- Mørk, M.; Andreasen, J.J.; Rasmussen, L.H.; Lip, G.Y.; Pedersen, S.; Bæk, R.; Jørgensen, M.M.; Kristensen, S.R. Elevated blood plasma levels of tissue factor-bearing extracellular vesicles in patients with atrial fibrillation. Thromb. Res. 2019, 173, 141–150. [Google Scholar] [CrossRef]
- Wu, C.-X.; Liu, Z.-F. Proteomic Profiling of Sweat Exosome Suggests its Involvement in Skin Immunity. J. Investig. Dermatol. 2018, 138, 89–97. [Google Scholar] [CrossRef]
- Somiya, M.; Yoshioka, Y.; Ochiya, T. Biocompatibility of highly purified bovine milk-derived extracellular vesicles. J. Extracell. Vesicles 2018, 7, 1440132. [Google Scholar] [CrossRef]
- Hyun, K.-A.; Gwak, H.; Lee, J.; Kwak, B.; Jung, H.-I. Salivary Exosome and Cell-Free DNA for Cancer Detection. Micromachines 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Raposo, G. Exosomes and extracellular vesicles: The path forward. Essays Biochem. 2018, 62, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Stoorvogel, W. Functional transfer of microRNA by exosomes. Blood 2012, 119, 646–648. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Rountree, R.B.; Mandl, S.J.; Nachtwey, J.M.; Dalpozzo, K.; Do, L.; Lombardo, J.R.; Schoonmaker, P.L.; Brinkmann, K.; Dirmeier, U.; Laus, R.; et al. Exosome Targeting of Tumor Antigens Expressed by Cancer Vaccines Can Improve Antigen Immunogenicity and Therapeutic Efficacy. Cancer Res. 2011, 71, 5235–5244. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Komuro, H.; Kawai-Harada, Y.; Aminova, S.; Pascual, N.; Malik, A.; Contag, C.H.; Harada, M. Engineering Extracellular Vesicles to Target Pancreatic Tissue In Vivo. Nanotheranostics 2021, 5, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Véron, P.; Segura, E.; Sugano, G.; Amigorena, S.; Théry, C. Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol. Dis. 2005, 35, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Zeelenberg, I.S.; Ostrowski, M.; Krumeich, S.; Bobrie, A.; Jancic, C.; Boissonnas, A.; Delcayre, A.; Le Pecq, J.-B.; Combadière, B.; Amigorena, S.; et al. Targeting Tumor Antigens to Secreted Membrane Vesicles In vivo Induces Efficient Antitumor Immune Responses. Cancer Res. 2008, 68, 1228–1235. [Google Scholar] [CrossRef]
- Hackel, B.J.; Neil, J.R.; White, F.M.; Wittrup, K. Epidermal growth factor receptor downregulation by small heterodimeric binding proteins. Protein Eng. Des. Sel. 2011, 25, 47–57. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef]
- Hackel, B.J.; Sathirachinda, A.; Gambhir, S.S. Designed hydrophilic and charge mutations of the fibronectin domain: Towards tailored protein biodistribution. Protein Eng. Des. Sel. 2012, 25, 639–648. [Google Scholar] [CrossRef]
- Hackel, B.J.; Kimura, R.H.; Gambhir, S.S. Use of64Cu-labeled Fibronectin Domain with EGFR-Overexpressing Tumor Xenograft: Molecular Imaging. Radiology 2012, 263, 179–188. [Google Scholar] [CrossRef]
- Woldring, D.; Holec, P.V.; Zhou, H.; Hackel, B.J. High-Throughput Ligand Discovery Reveals a Sitewise Gradient of Diversity in Broadly Evolved Hydrophilic Fibronectin Domains. PLoS ONE 2015, 10, e0138956. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kim, M. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Wiklander, P.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Dsc, A.H.V.S.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef]
- Sedlik, C.; Vigneron, J.; Torrieri-Dramard, L.; Pitoiset, F.; Denizeau, J.; Chesneau, C.; De La Rochère, P.; Lantz, O.; Thery, C.; Bellier, B. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J. Extracell. Vesicles 2014, 3, 24646. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Gitz-Francois, J.J.J.M.; Schiffelers, R.M.; Vader, P. Recombinant phosphatidylserine-binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug-and-play approach. Nanoscale 2018, 10, 2413–2426. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Wei, J.; Glass, O.K.; Guo, H.; Lei, G.; Yang, X.-Y.; Osada, T.; Hobeika, A.; Delcayre, A.; Le Pecq, J.-B.; et al. Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361–9367. [Google Scholar] [CrossRef]
- Wang, J.H.; Forterre, A.V.; Zhao, J.; Frimannsson, D.O.; Delcayre, A.; Antes, T.J.; Efron, B.; Jeffrey, S.S.; Pegram, M.D.; Matin, A.C. Anti-HER2 scFv-Directed Extracellular Vesicle-Mediated mRNA-Based Gene Delivery Inhibits Growth of HER2-Positive Human Breast Tumor Xenografts by Prodrug Activation. Mol. Cancer Ther. 2018, 17, 1133–1142. [Google Scholar] [CrossRef]
- Wan, S.; Zhang, L.; Wang, S.; Liu, Y.; Wu, C.; Cui, C.; Sun, H.; Shi, M.; Jiang, Y.; Li, L.; et al. Molecular Recognition-Based DNA Nanoassemblies on the Surfaces of Nanosized Exosomes. J. Am. Chem. Soc. 2017, 139, 5289–5292. [Google Scholar] [CrossRef]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Kato, K.; Yamashita, T.; Imai, T.; Saji, H.; Takakura, Y. Quantitative Analysis of Tissue Distribution of the B16BL6-Derived Exosomes Using a Streptavidin-Lactadherin Fusion Protein and Iodine-125-Labeled Biotin Derivative After Intravenous Injection in Mice. J. Pharm. Sci. 2015, 104, 705–713. [Google Scholar] [CrossRef]
- Stern, L.; Schrack, I.A.; Johnson, S.M.; Deshpande, A.; Bennett, N.R.; Harasymiw, L.A.; Gardner, M.K.; Hackel, B.J. Geometry and expression enhance enrichment of functional yeast-displayed ligands via cell panning. Biotechnol. Bioeng. 2016, 113, 2328–2341. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Saltik, M.; Lehrmann, H.; Killisch, I.; Mautner, V.; Lamm, G.; Christofori, G.; Cotten, M. Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene Ther. 1997, 4, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Ritter, S.C.; Hackel, B.J. Validation and Stabilization of a Prophage Lysin of Clostridium perfringens by Using Yeast Surface Display and Coevolutionary Models. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, G.A.; Fernandes, A.V.; Sundararaghavan, H.G.; Shreiber, D.I. Positively and Negatively Modulating Cell Adhesion to Type I Collagen Via Peptide Grafting. Tissue Eng. Part A 2011, 17, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; De Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol. Ther.-Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Liang, G.; Kan, S.; Zhu, Y.; Feng, S.; Feng, W.; Gao, S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomed. 2018, 13, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; Le Pecq, J.-B. Exosome Display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.-I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef]
- Campos-Silva, C.; Suárez, H.; Acevedo, R.J.; Linares-Espinós, E.; Martinez-Piñeiro, L.; Yáñez-Mó, M.; Valés-Gómez, M. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Im, E.-J.; Lee, C.-H.; Moon, P.-G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.-C.; Jee, J.-G.; Bae, J.-S.; Kwon, T.-K.; et al. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin. Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef]
- Miki, Y.; Ono, K.; Hata, S.; Suzuki, T.; Kumamoto, H.; Sasano, H. The advantages of co-culture over mono cell culture in simulating in vivo environment. J. Steroid Biochem. Mol. Biol. 2012, 131, 68–75. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komuro, H.; Aminova, S.; Lauro, K.; Woldring, D.; Harada, M. Design and Evaluation of Engineered Extracellular Vesicle (EV)-Based Targeting for EGFR-Overexpressing Tumor Cells Using Monobody Display. Bioengineering 2022, 9, 56. https://doi.org/10.3390/bioengineering9020056
Komuro H, Aminova S, Lauro K, Woldring D, Harada M. Design and Evaluation of Engineered Extracellular Vesicle (EV)-Based Targeting for EGFR-Overexpressing Tumor Cells Using Monobody Display. Bioengineering. 2022; 9(2):56. https://doi.org/10.3390/bioengineering9020056
Chicago/Turabian StyleKomuro, Hiroaki, Shakhlo Aminova, Katherine Lauro, Daniel Woldring, and Masako Harada. 2022. "Design and Evaluation of Engineered Extracellular Vesicle (EV)-Based Targeting for EGFR-Overexpressing Tumor Cells Using Monobody Display" Bioengineering 9, no. 2: 56. https://doi.org/10.3390/bioengineering9020056
APA StyleKomuro, H., Aminova, S., Lauro, K., Woldring, D., & Harada, M. (2022). Design and Evaluation of Engineered Extracellular Vesicle (EV)-Based Targeting for EGFR-Overexpressing Tumor Cells Using Monobody Display. Bioengineering, 9(2), 56. https://doi.org/10.3390/bioengineering9020056