
Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells

 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Osteogenic Differentiation
2.2. Alkaline Phosphatase Assay
2.3. Alizarin Red Staining
2.4. Gene Expression/q-PCR Analysis
2.5. Cell Culture and RNA Isolation for RNA-Sequencing
2.6. Cell Culture and Sample Processing for Intracellular Metabolomics Data
2.7. Glucose and Lactate Measurements
2.8. Glutamine, Glutamic Acid, and Ammonia Measurements
2.9. RNA-Sequencing Data Analysis
2.9.1. Preparation of RNA-Seq Libraries
2.9.2. Sequencing
2.9.3. Gene Expression Quantification
2.9.4. Differential Gene Expression Analysis
2.9.5. Gene Set Ontology Enrichment Analysis
2.9.6. Regulatory Influence Inference Algorithm
2.9.7. TF–TF Regulatory Influence Network Visualization
2.9.8. Code Availability
2.10. Intracellular Labelled and Unlabeled Metabolite Extraction
2.11. BCA Protein Assay
2.12. UPLC-MS Set Up and Run Configuration
2.13. Metabolomics Data Normalization and Processing
2.13.1. Extracellular Metabolomic Data
2.13.2. Intracellular Unlabeled Metabolomic Data
2.13.3. Mummichog Pathway and Network Analysis
2.13.4. Intracellular Labeled Metabolomic Data
3. Results
3.1. Verification of Osteogenic Differentiation
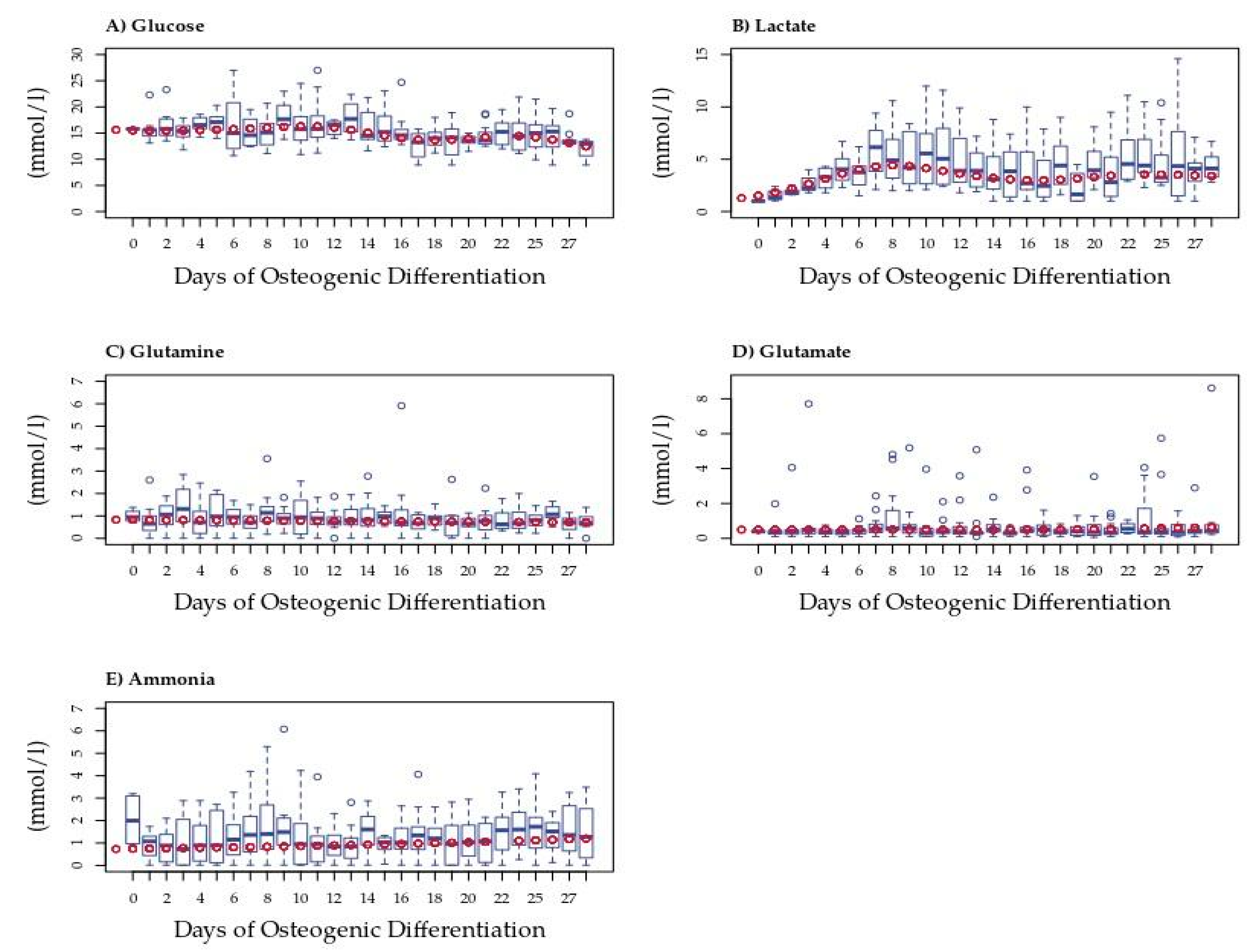
3.2. Lactate Secretion Changes during Osteogenic Differentiation
3.3. The Utilization and Production of Essential Metabolites Are Indicative of Three Stages of Osteogenic Differentiation
3.4. 13C-Glucose Mean Label Enrichment Suggests an Anaerobic Switch following Day 9 of Osteogenic Differentiation
3.5. Mitochondrial Activity Increases as Differentiation Progresses and Is Significantly Different between Suggested Phases
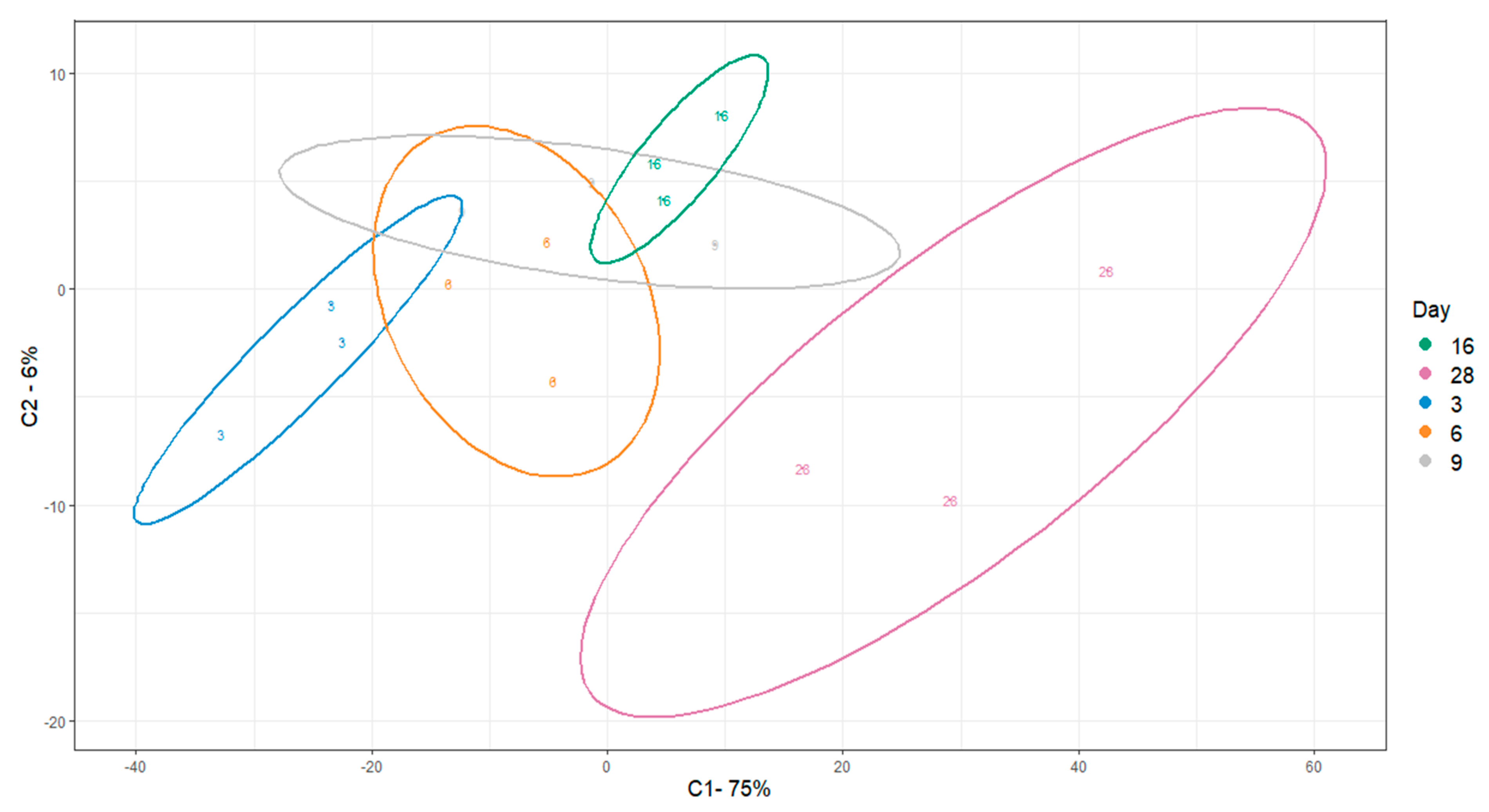
3.6. Intracellular Metabolomics Validate Metabolic Stages of Osteogenic Differentiation
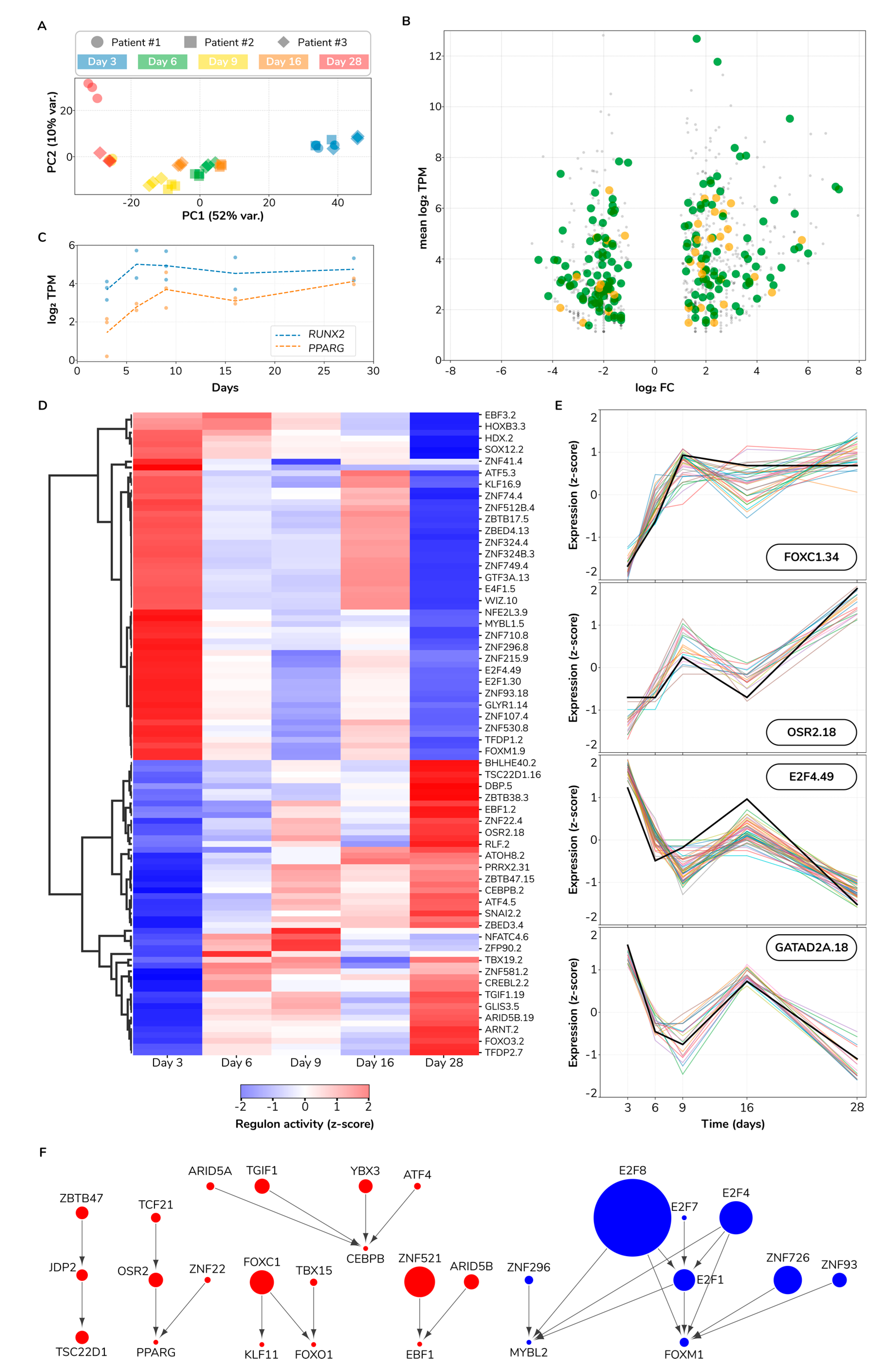
3.7. Gene Expression Analysis Indicates Major Changes in Cellular Function during Osteogenic Differentiation
3.8. Regulatory Influence Inference Reveals a Cell State Switch
4. Discussion
4.1. Increase in Mitochondrial Activity following Day 9
4.2. Increased Glycolysis followed by Decreased PPP Flux as Differentiation Progresses
4.3. Temporal Changes in Glutaminolysis and TCA-Cycle Activity
4.4. Unexpected Changes in Lipolysis Suggest Further Investigation
4.5. Upregulated Lipid Metabolism and Downregulated Cell Cycle-Related Anabolic Activity Observed via DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campana, V.; Milano, G.; Pagano, E.D.; Barba, M.; Cicione, C.; Salonna, G.; Lattanzi, W.; Logroscino, G. Bone substitutes in orthopaedic surgery: From basic science to clinical practice. J. Mater. Sci. Mater. Med. 2014, 25, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, E.; Cadossi, M.; Tedesco, G.; Capra, P.; Calamelli, C.; Shehu, A.; Giannini, S. Autograft, allograft and bone substitutes in reconstructive orthopedic surgery. Aging Clin. Exp. Res. 2013, 25, 101–103. [Google Scholar] [CrossRef]
- Bone Grafts & Substitutes Market Size, Share|Industry Report, 2026. Available online: https://www.grandviewresearch.com/industry-analysis/bone-grafts-substitutes-market (accessed on 23 March 2021).
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef]
- Schemitsch, E.H.; Nauth, A. Stem cells for the repair and regeneration of bone. Indian J. Orthop. 2012, 46, 19–21. [Google Scholar] [CrossRef]
- Neman, J.; Hambrecht, A.; Jandial, R.; Cadry, C. Stem cell-mediated osteogenesis: Therapeutic potential for bone tissue engineering. Biol. Targets Ther. 2012, 6, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Vaishya, R.; Chauhan, M.; Vaish, A. Bone cement. J. Clin. Orthop. Trauma 2013, 4, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Gazit, Z.; Pelled, G.; Sheyn, D.; Kimelman, N.; Gazit, D. Mesenchymal Stem Cells. In Essentials of Stem Cell Biology, 3rd ed.; Lanza, R., Atala, A., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 255–266. [Google Scholar]
- Deng, Z.-L.; Sharff, K.; Tang, N.; Song, W.-X.; Luo, J.; Luo, X.; Chen, J.; Bennett, E.; Reid, R.; Manning, D.; et al. Regulation of osteogenic differentiation during skeletal development. Front. Biosci. 2008, 13, 2001–2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shih, Y.V.; Kuo, T.K.; Lee, O.K.; Wei, Y. Coordinated Changes of Mitochondrial Biogenesis and Antioxidant Enzymes During Osteogenic Differentiation of Human Mesenchymal Stem Cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef]
- Pattappa, G.; Heywood, H.K.; De Bruijn, J.D.; Lee, D.A. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J. Cell. Physiol. 2011, 226, 2562–2570. [Google Scholar] [CrossRef]
- Shum, L.; White, N.S.; Mills, B.N.; Bentley, K.L.D.M.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells During Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Forni, M.F.; Peloggia, J.; Trudeau, K.; Shirihai, O.; Kowaltowski, A.J. Murine Mesenchymal Stem Cell Commitment to Differentiation Is Regulated by Mitochondrial Dynamics. Stem Cells 2015, 34, 743–755. [Google Scholar] [CrossRef]
- Meleshina, A.V.; Dudenkova, V.V.; Shirmanova, M.V.; Shcheslavskiy, V.I.; Becker, W.; Bystrova, A.S.; Cherkasova, E.I.; Zagaynova, E.V. Probing metabolic states of differentiating stem cells using two-photon FLIM. Sci. Rep. 2016, 6, 21853. [Google Scholar] [CrossRef]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.-X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef]
- Huang, T.; Liu, R.; Fu, X.; Yao, D.; Yang, M.; Liu, Q.; Lu, W.W.; Wu, C.; Guan, M. Aging Reduces an ERRalpha-Directed Mitochondrial Glutaminase Expression Suppressing Glutamine Anaplerosis and Osteogenic Differentiation of Mesenchymal Stem Cells. Stem Cells 2016, 35, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Hutchison, J.D.; Crockett, J.C. Absence of Glutamine Supplementation Prevents Differentiation of Murine Calvarial Osteoblasts to a Mineralizing Phenotype. Calcif. Tissue Int. 2011, 89, 472–482. [Google Scholar] [CrossRef]
- Zhao, Q.; Ren, H.; Han, Z. Mesenchymal stem cells: Immunomodulatory capability and clinical potential in immune diseases. J. Cell. Immunother. 2016, 2, 3–20. [Google Scholar] [CrossRef]
- Simon, L.; Edelstein, L.; Nagalla, S.; Woodley, A.B.; Chen, E.S.; Kong, X.; Ma, L.; Fortina, P.; Kunapuli, S.; Holinstat, M.; et al. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood 2014, 123, e37–e45. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Gonzalez, G.A.P.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.; Shu, S.; Marshall, B.; Lewis, S.; The AmiGO Hub; The Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2008, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 October 2020).
- Wood, S.; Scheipl, F. Package ‘gamm4′: Generalized Additive Mixed Models Using ‘mgcv’ and ‘lme4.’ 3 April 2020. Available online: https://cran.r-project.org/web/packages/gamm4/gamm4.pdf (accessed on 1 July 2021).
- James, N.A.; Matteson, D.S. ecp: An R Package for Nonparametric Multiple Change Point Analysis of Multivariate Data ArXiv13093295 Stat, Nov. Available online: http://arxiv.org/abs/1309.3295 (accessed on 10 October 2020).
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting Network Activity from High Throughput Metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef]
- IsoCor: Isotope Correction for Mass Spectrometry Labeling Experiment—IsoCor 2.2.0 Documentation. Available online: https://isocor.readthedocs.io/en/latest/ (accessed on 23 February 2021).
- Bruderer, M.; Richards, R.G.; Alini, M.; Stoddart, M.J. Role and regulation of RUNX2 in osteogenesis. Eur. Cell. Mater. 2014, 28, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Golub, E.; Boesze-Battaglia, K. The role of alkaline phosphatase in mineralization. Curr. Opin. Orthop. 2007, 18, 444–448. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Kawai, S.; Kato, T.; Ooshima, T.; Amano, A. Odd-skipped related 1 gene expression is regulated by Runx2 and Ikzf1 transcription factors. Gene 2008, 426, 81–90. [Google Scholar] [CrossRef]
- Stricker, S.; Mathia, S.; Haupt, J.; Seemann, P.; Meier, J.; Mundlos, S. Odd-Skipped Related Genes Regulate Differentiation of Embryonic Limb Mesenchyme and Bone Marrow Mesenchymal Stromal Cells. Stem Cells Dev. 2012, 21, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Pricola, K.L.; Kuhn, N.Z.; Haleem-Smith, H.; Song, Y.; Tuan, R.S. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. J. Cell. Biochem. 2009, 108, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Weng, Y.; Yuan, T.; Zhang, H.; Bai, H.; Li, B.; Yang, D.; Zhang, R.; He, F.; Yan, S.; et al. CXCL12/CXCR4 Signal Axis Plays an Important Role in Mediating Bone Morphogenetic Protein 9-induced Osteogenic Differentiation of Mesenchymal Stem Cells. Int. J. Med. Sci. 2013, 10, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a Candidate Gene for Cleidocranial Dysplasia Syndrome, Is Essential for Osteoblast Differentiation and Bone Development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ Is Required for the Differentiation of Adipose Tissue In Vivo and In Vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Stechschulte, L.A.; Lecka-Czernik, B. Reciprocal Regulation of PPARγ and RUNX2 Activities in Marrow Mesenchymal Stem Cells: Fine Balance between p38 MAPK and Protein Phosphatase. Curr. Mol. Biol. Rep. 2017, 3, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Umek, R.M.; McKnight, S.L. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991, 5, 1538–1552. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-Regulation of C/EBPα and PPARγ Controls the Transcriptional Pathway of Adipogenesis and Insulin Sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Tominaga, H.; Maeda, S.; Hayashi, M.; Takeda, S.; Akira, S.; Komiya, S.; Nakamura, T.; Akiyama, H.; Imamura, T. CCAAT/Enhancer-binding Protein β Promotes Osteoblast Differentiation by Enhancing Runx2 Activity with ATF4. Mol. Biol. Cell 2008, 19, 5373–5386. [Google Scholar] [CrossRef][Green Version]
- Meyer, M.B.; Benkusky, N.A.; Sen, B.; Rubin, J.; Pike, J.W. Epigenetic Plasticity Drives Adipogenic and Osteogenic Differentiation of Marrow-derived Mesenchymal Stem Cells. J. Biol. Chem. 2016, 291, 17829–17847. [Google Scholar] [CrossRef] [PubMed]
- Saint-Antoine, M.M.; Singh, A. Network inference in systems biology: Recent developments, challenges, and applications. Curr. Opin. Biotechnol. 2020, 63, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, E.; Aloisio, A.; Codispoti, B.; Nappo, G.; Scicchitano, S.; Lucchino, V.; Montalcini, Y.; Camarotti, A.; Galasso, O.; Greco, M.; et al. ZNF521 Has an Inhibitory Effect on the Adipogenic Differentiation of Human Adipose-Derived Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2018, 14, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, F.; Lavy, R.; Penner-Chea, J.; Berry, F.B. Initiation of Early Osteoblast Differentiation Events through the Direct Transcriptional Regulation of Msx2 by FOXC1. PLoS ONE 2012, 7, e49095. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Sun, Y.; Zhang, Z. Identification of key transcription factors—Gene regulatory network related with osteogenic differentiation of human mesenchymal stem cells based on transcription factor prognosis system. Exp. Ther. Med. 2019, 17, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Gasser, A.; Bolamperti, S.; Maeda, M.; Matthies, L.; Jähn, K.; Long, C.L.; Schlüter, H.; Kwiatkowski, M.; Saini, V.; et al. TG-interacting factor 1 (Tgif1)-deficiency attenuates bone remodeling and blunts the anabolic response to parathyroid hormone. Nat. Commun. 2019, 10, 1354. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Yamauchi, M.; Wakisaka, S.; Ooshima, T.; Amano, A. Zinc-Finger Transcription Factor Odd-Skipped Related 2 Is One of the Regulators in Osteoblast Proliferation and Bone Formation. J. Bone Miner. Res. 2007, 22, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs Regulate Adipocyte Differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef]
- Flowers, S.; Xu, F.; Moran, E. Cooperative Activation of Tissue-Specific Genes by pRB and E2F1. Cancer Res. 2013, 73, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, M.; Carpenter, S.R.; Lenton, T.M.; Bascompte, J.; Brock, W.; Dakos, V.; van de Koppel, J.; van de Leemput, I.A.; Levin, S.A.; van Nes, E.H.; et al. Anticipating Critical Transitions. Science 2012, 338, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Moris, N.; Pina, C.; Arias, A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016, 17, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef]
- Hanna, H.; Mir, L.M.; Andre, F.M. In vitro osteoblastic differentiation of mesenchymal stem cells generates cell layers with distinct properties. Stem Cell Res. Ther. 2018, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.O.; Eliseev, R.A. Energy Metabolism During Osteogenic Differentiation: The Role of Akt. Stem Cells Dev. 2021, 30, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Dobson, P.F.; Dennis, E.P.; Hipps, D.; Reeve, A.; Laude, A.; Bradshaw, C.; Stamp, C.; Smith, A.; Deehan, D.J.; Turnbull, D.M.; et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci. Rep. 2020, 10, 11643. [Google Scholar] [CrossRef]
- Maneiro, E.; Martín, M.Á.; De Andres, M.C.; López-Armada, M.J.; Fernández-Sueiro, J.L.; Del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003, 48, 700–708. [Google Scholar] [CrossRef]
- Zhang, D.; Yan, B.; Yu, S.; Zhang, C.; Wang, B.; Wang, Y.; Wang, J.; Yuan, Z.; Zhang, L.; Pan, J. Coenzyme Q10 Inhibits the Aging of Mesenchymal Stem Cells Induced by D-Galactose through Akt/mTOR Signaling. Oxidative Med. Cell. Longev. 2015, 2015, 867293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Cui, C.; Yu, M.; Li, X.; Wang, L.; Chen, X.; Lin, Y. Coenzyme Q10 promotes osteoblast proliferation and differentiation and protects against ovariectomy-induced osteoporosis. Mol. Med. Rep. 2017, 17, 400–407. [Google Scholar] [CrossRef]
- Luo, K.; Yu, J.H.; Quan, Y.; Shin, Y.J.; Lee, K.E.; Kim, H.L.; Ko, E.J.; Chung, B.H.; Lim, S.W.; Yang, C.W. Therapeutic potential of coenzyme Q10 in mitochondrial dysfunction during tacrolimus-induced beta cell injury. Sci. Rep. 2019, 9, 7995. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.-Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 Supplementation Activates Mitochondria Functions to Decelerate Senescence in Senescence-Accelerated Mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Toscani, D.; Marchica, V.; Taurino, G.; Costa, F.; Bianchi, M.; Andreoli, R.; Franceschi, V.; Storti, P.; Burroughs-Garcia, J.; et al. Myeloma Cells Deplete Bone Marrow Glutamine and Inhibit Osteoblast Differentiation Limiting Asparagine Availability. Cancers 2020, 12, 3267. [Google Scholar] [CrossRef]
- Stegen, S.; Devignes, C.; Torrekens, S.; Van Looveren, R.; Carmeliet, P.; Carmeliet, G. Glutamine Metabolism in Osteoprogenitors Is Required for Bone Mass Accrual and PTH -Induced Bone Anabolism in Male Mice. J. Bone Miner. Res. 2021, 36, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Alekos, N.S.; Moorer, M.C.; Riddle, R.C. Dual Effects of Lipid Metabolism on Osteoblast Function. Front. Endocrinol. 2020, 11, 578194. [Google Scholar] [CrossRef] [PubMed]
- How Agilent Seahorse XF Analyzers Work|Agilent. Available online: https://www.agilent.com/en/products/cell-analysis/how-seahorse-xf-analyzers-work (accessed on 13 January 2021).
- Nicholas, D.; Proctor, E.A.; Raval, F.M.; Ip, B.C.; Habib, C.; Ritou, E.; Grammatopoulos, T.N.; Steenkamp, D.; Dooms, H.; Apovian, C.M.; et al. Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS ONE 2017, 12, e0170975. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Glucose/Lactate | Glutamine/Glutamate | Glutamine/Ammonia |
|---|---|---|---|
| 1 (Days 1–4) | −0.209 | −2.620 | −0.444 |
| 2 (Days 5–15) | 0.088 * | −38.299 | −0.374 |
| 3 (Days 16–28) | −0.908 | −1.843 | −0.284 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigmarsdottir, T.B.; McGarrity, S.; de Lomana, A.L.G.; Kotronoulas, A.; Sigurdsson, S.; Yurkovich, J.T.; Rolfsson, O.; Sigurjonsson, O.E. Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells. Bioengineering 2021, 8, 208. https://doi.org/10.3390/bioengineering8120208
Sigmarsdottir TB, McGarrity S, de Lomana ALG, Kotronoulas A, Sigurdsson S, Yurkovich JT, Rolfsson O, Sigurjonsson OE. Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells. Bioengineering. 2021; 8(12):208. https://doi.org/10.3390/bioengineering8120208
Chicago/Turabian StyleSigmarsdottir, Thora Bjorg, Sarah McGarrity, Adrián López García de Lomana, Aristotelis Kotronoulas, Snaevar Sigurdsson, James T. Yurkovich, Ottar Rolfsson, and Olafur Eysteinn Sigurjonsson. 2021. "Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells" Bioengineering 8, no. 12: 208. https://doi.org/10.3390/bioengineering8120208
APA StyleSigmarsdottir, T. B., McGarrity, S., de Lomana, A. L. G., Kotronoulas, A., Sigurdsson, S., Yurkovich, J. T., Rolfsson, O., & Sigurjonsson, O. E. (2021). Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells. Bioengineering, 8(12), 208. https://doi.org/10.3390/bioengineering8120208