Influence of Pathogens and Mechanical Stimuli in Inflammation


Abstract
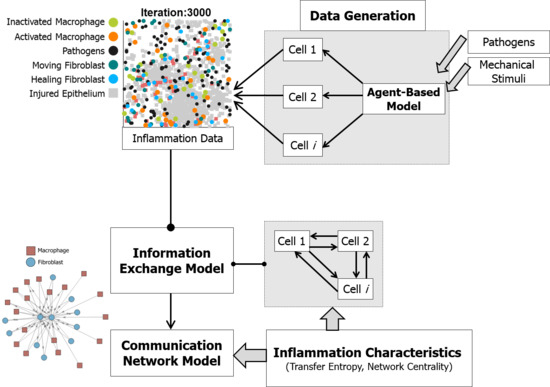
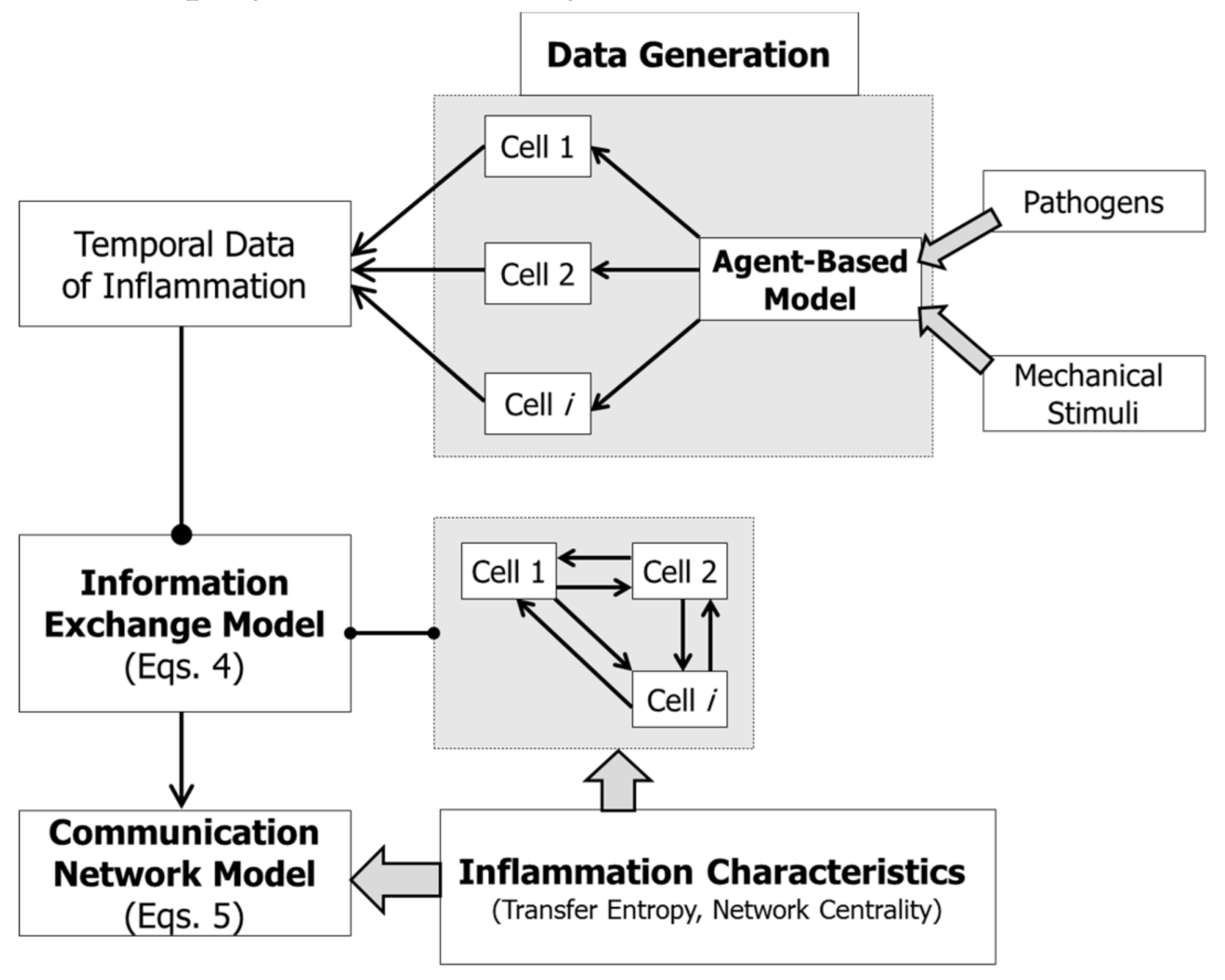
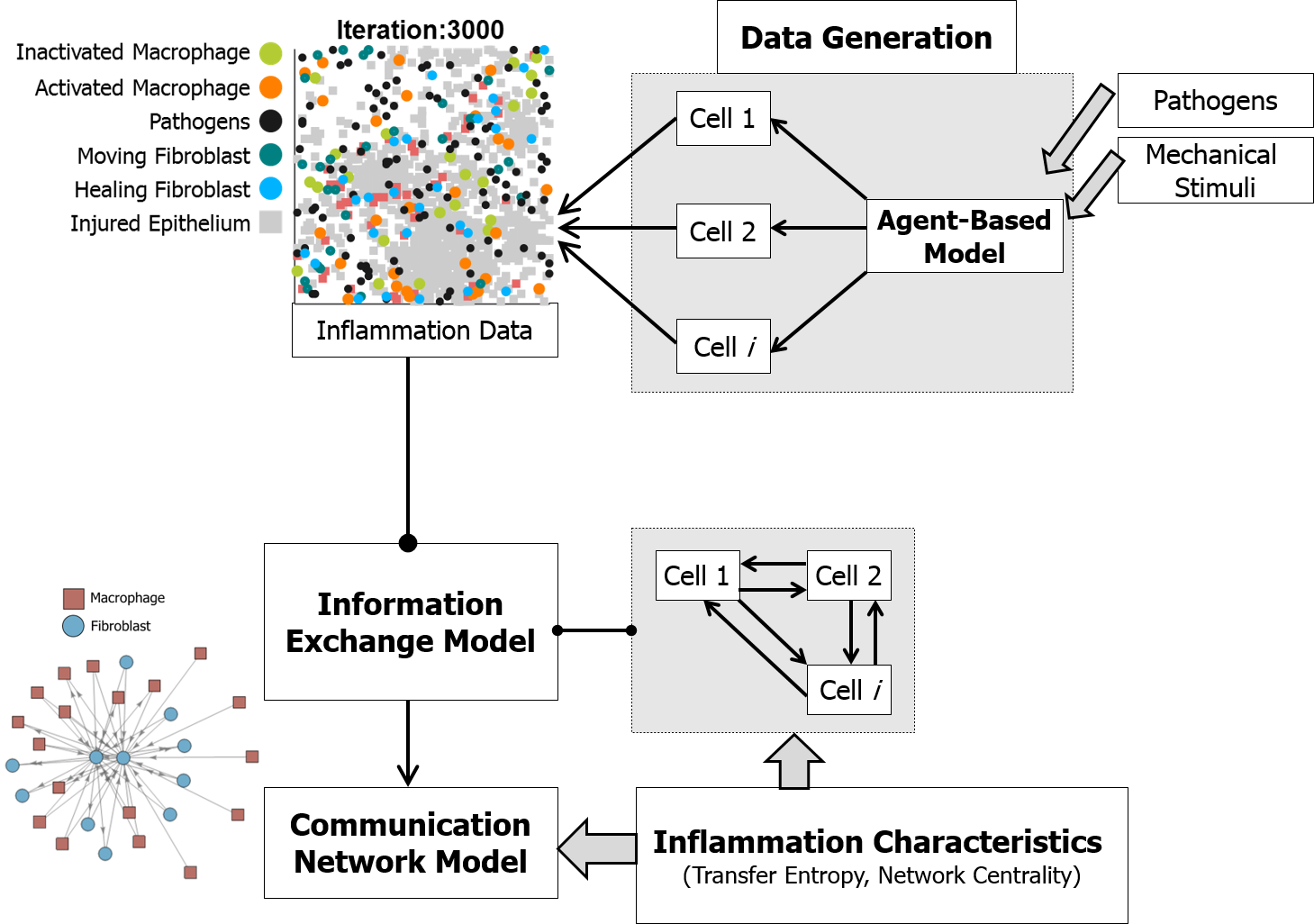{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
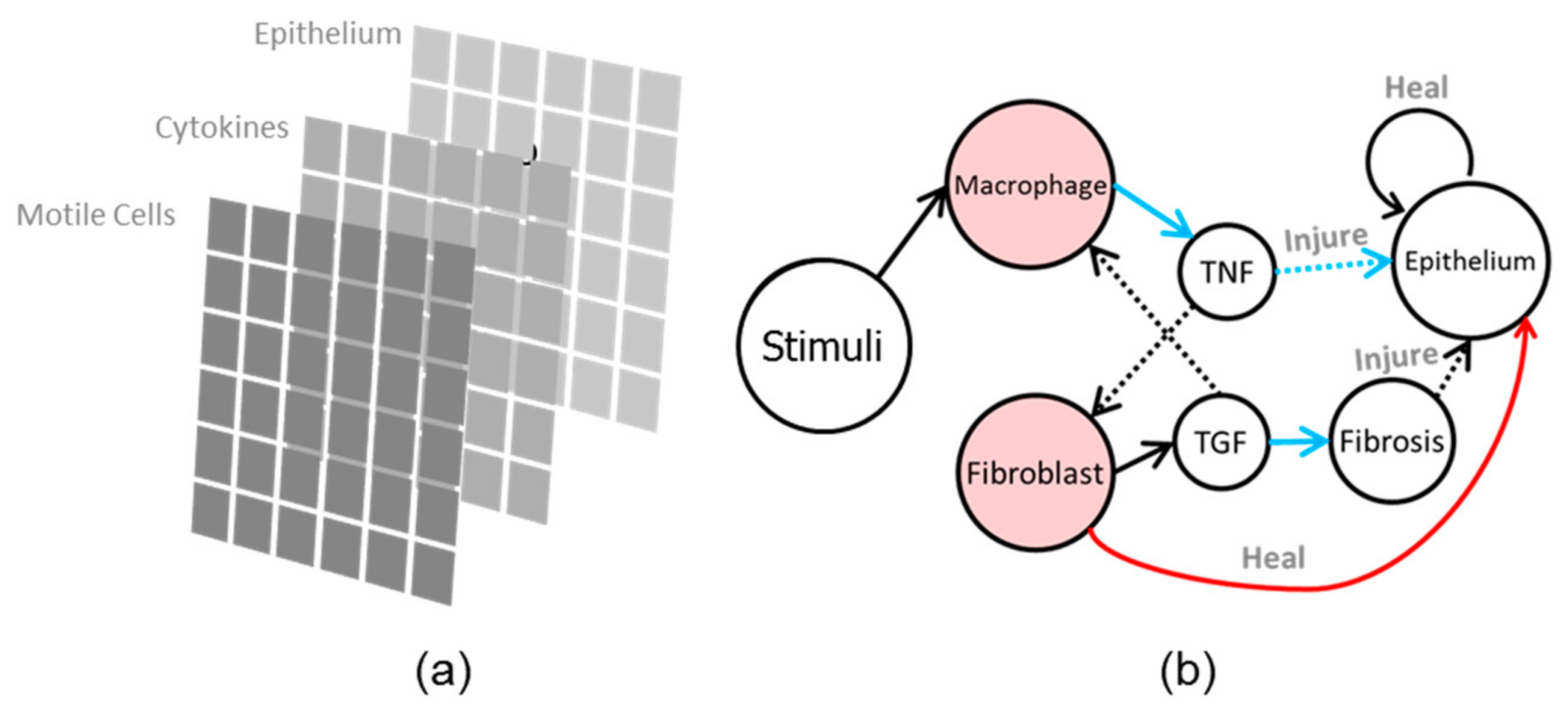
2.1. Physiology of Inflammation
2.2. Agent-Based Model for Inflammation
2.2.1. Epithelial Cells
2.2.2. Motile Cells
2.2.3. Cytokines Diffusion
2.2.4. Pathogens and Strain
2.3. Information Exchange Model
2.4. Communication Network Model
3. Results and Discussion
3.1. Data Generation
3.2. Comparison of Inflammation Time Course
3.3. Dynamics of Information Exchange between Macrophage and Fibroblast
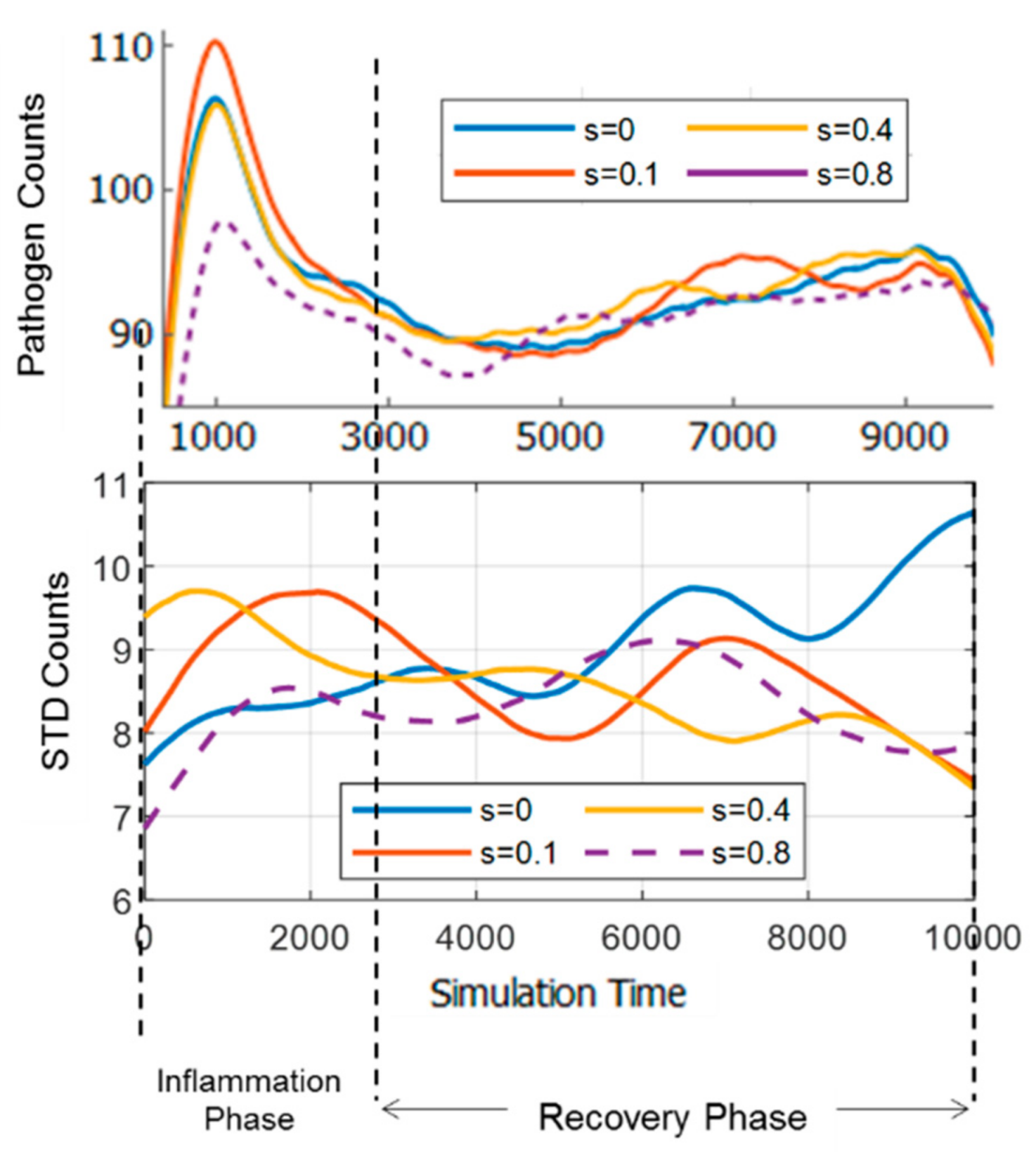
3.4. Effect of Strain and Pathogen to Cell’s Information Exchange
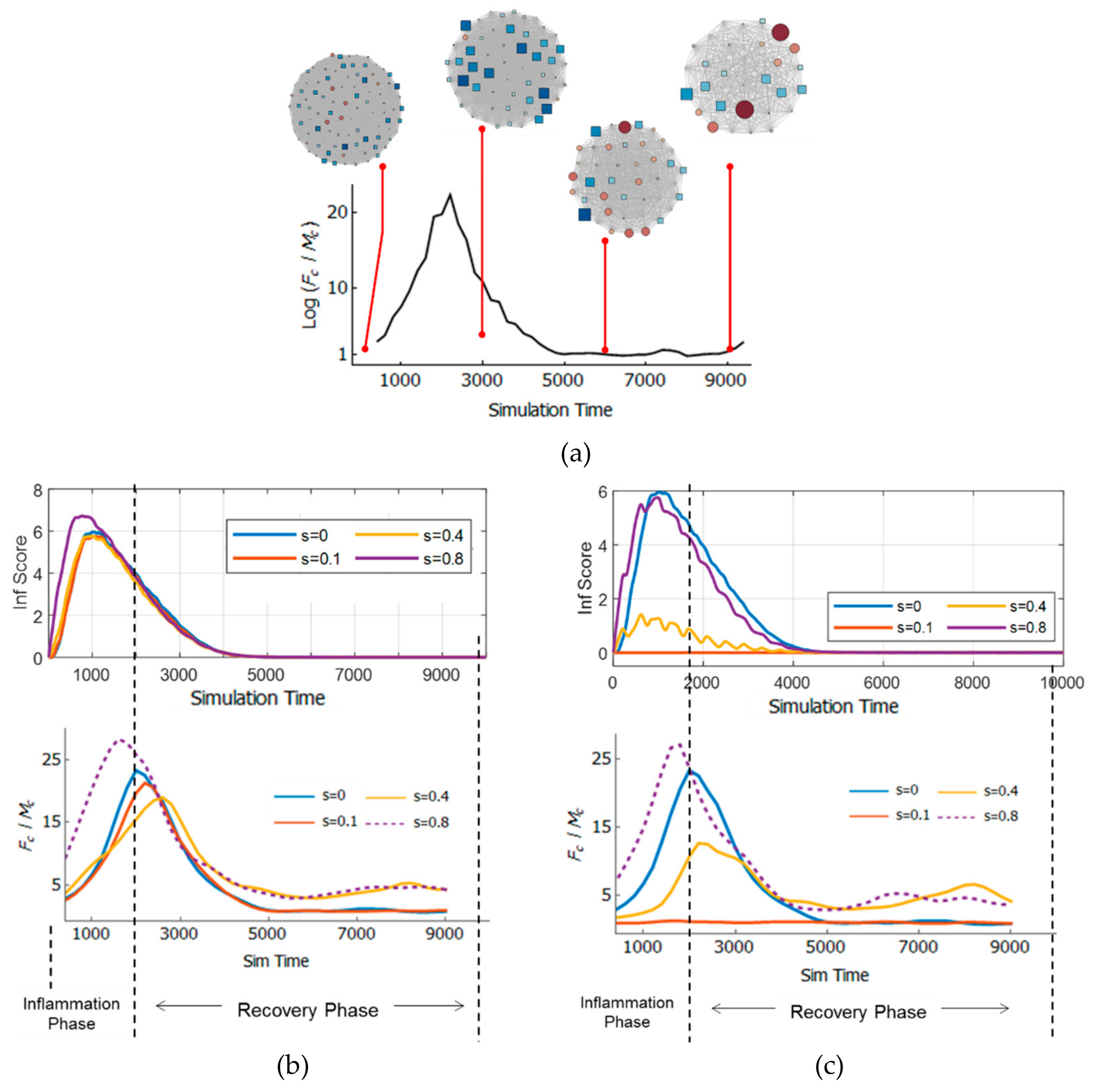
3.5. Resilience of the Innate Immune System
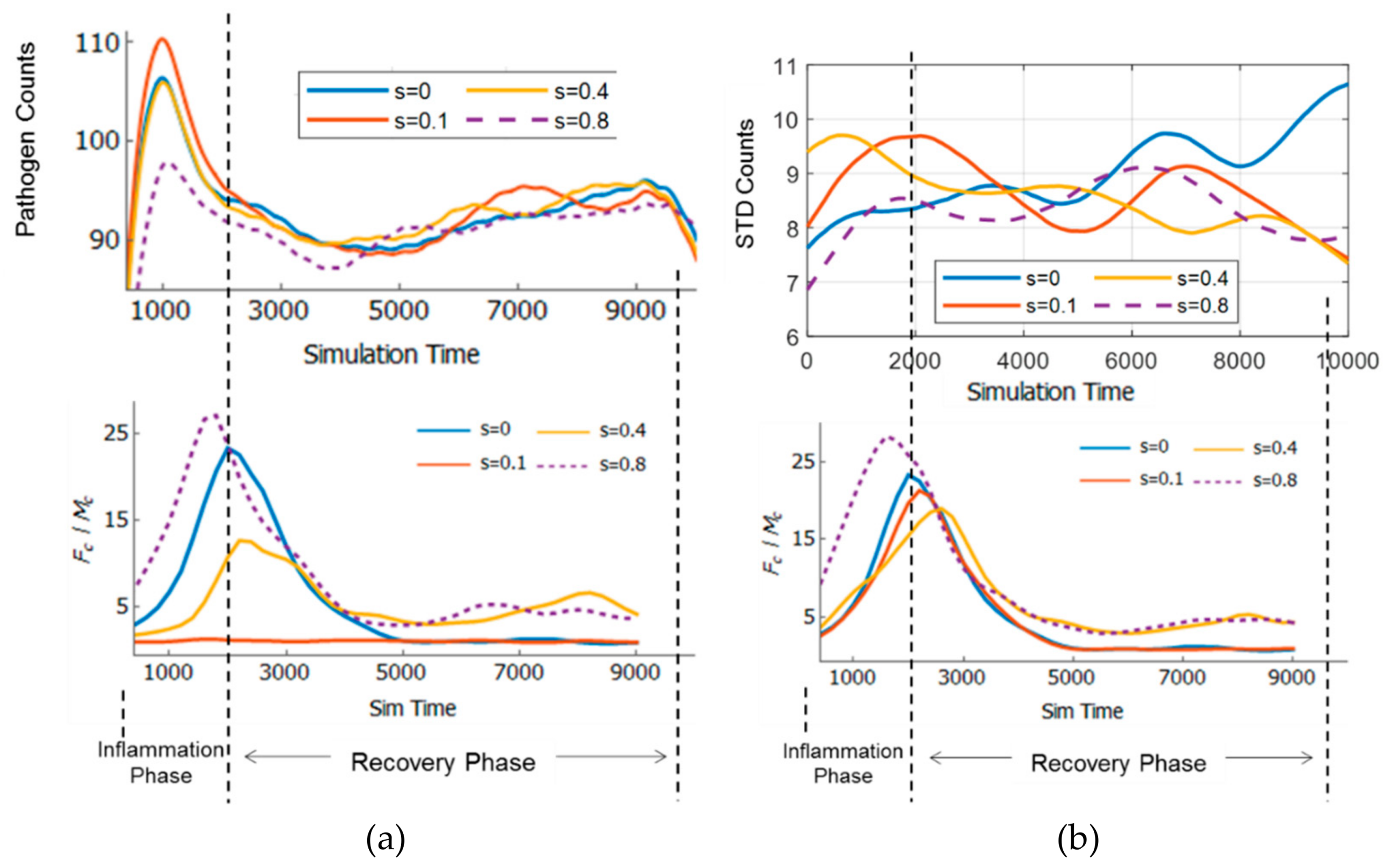
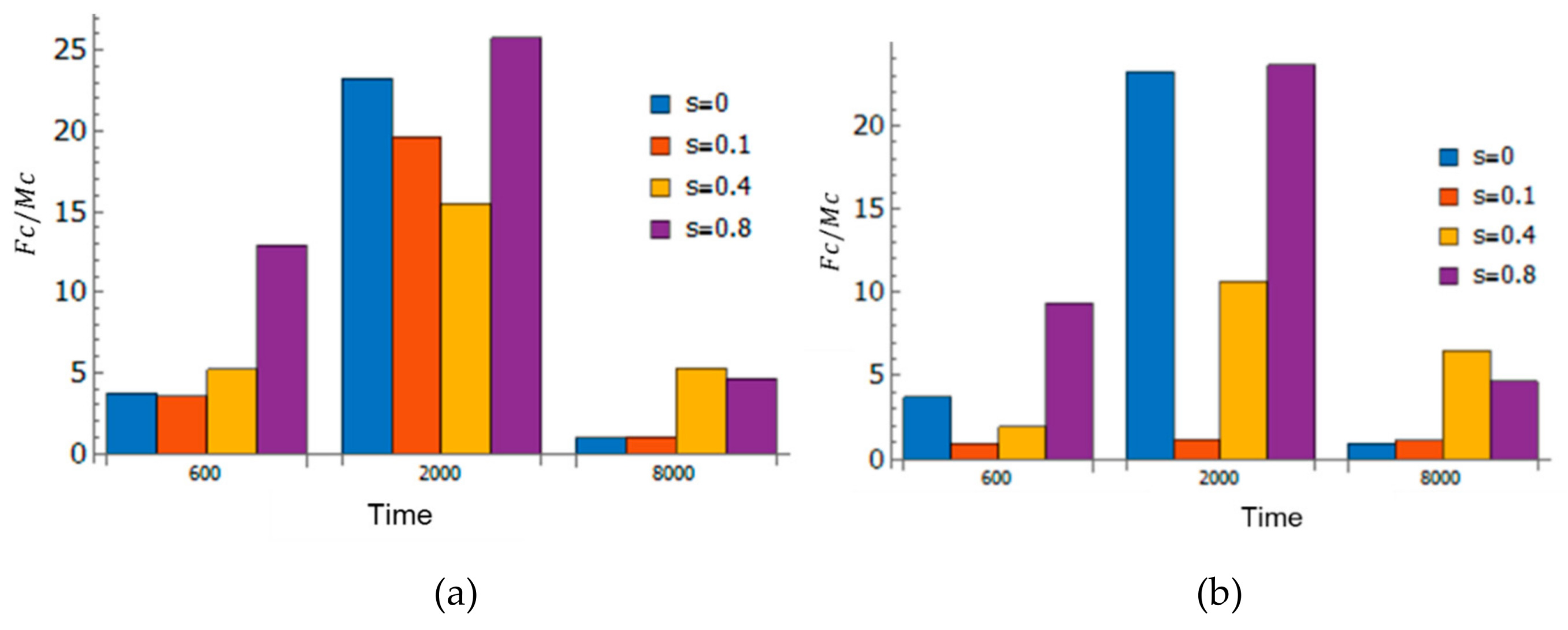
3.6. Adaptivity of Innate Immune System
3.7. Limitations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi Narendra, B.; Eshvendar Reddy, K.; Shantikumar, S.; Ramakrishna, S. Immune system: A double-edged sword in cancer. Inflamm. Res. 2013, 62, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Tranquillo, R.T.; Murray, J.D. Continuum model of fibroblast-driven wound contraction: Inflammation-mediation. J. Theor. Biol. 1992, 158, 135–172. [Google Scholar] [CrossRef]
- Javierre, E.; Moreo, P.; Doblaré, M.; García-Aznar, J.M. Numerical modeling of a mechano-chemical theory for wound contraction analysis. Int. J. Solids Struct. 2009, 46, 3597–3606. [Google Scholar] [CrossRef][Green Version]
- Reynolds, A.; Koombua, K.; Pidaparti, R.M.; Ward, K.R. Cellular Automata Modeling of Pulmonary Inflammation. Mol. Cell. Biomech. 2012, 9, 141–156. [Google Scholar] [PubMed]
- Brown, B.N.; Price, I.M.; Toapanta, F.R.; DeAlmeida, D.R.; Wiley, C.A.; Ross, T.M.; Oury, T.D.; Vodovotz, Y. An agent-based model of inflammation and fibrosis following particulate exposure in the lung. Math. Biosci. 2011, 231, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Wellman, T.J.; Mondoñedo, J.R.; Davis, G.S.; Bates, J.H.T.; Suki, B. Topographic distribution of idiopathic pulmonary fibrosis: A hybrid physics-and agent-based model. Physiol. Meas. 2018, 39, 64007. [Google Scholar] [CrossRef] [PubMed]
- Dutta-Moscato, J.; Solovyev, A.; Mi, Q.; Nishikawa, T.; Soto-Gutierrez, A.; Fox, I.J.; Vodovotz, Y. A Multiscale Agent-Based in silico Model of Liver Fibrosis Progression. Front. Bioeng. Biotechnol. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Uhlig, U. Pharmacological interventions in ventilator-induced lung injury. Trends Pharmacol. Sci. 2004, 25, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y. Computational modelling of the inflammatory response in trauma, sepsis and wound healing: implications for modelling resilience. Interface Focus 2014, 4, 20140004. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Aguda, B.D.; Rath, B.; Agarwal, S. Biomechanical thresholds regulate inflammation through the NF-κB pathway: Experiments and modeling. PLoS ONE 2009, 4, e5262. [Google Scholar] [CrossRef] [PubMed]
- Rath, P.C.; Aggarwal, B.B. TNF-induced signaling in apoptosis. J. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Kelso, A. Cytokines: Principles and prospects. Immunol. Cell Biol. 1998, 76, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Arnold, R. TNF-α induces TGFβ1 - expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell Mol. Med. 2010, 13, 1866–1876. [Google Scholar] [CrossRef]
- Poujade, M.; Grasland-Mongrain, E.; Hertzog, A.; Jouanneau, J.; Chavrier, P.; Ladoux, B.; Buguin, A.; Silberzan, P. Collective migration of an epithelial monolayer in response to a model wound. Proc. Natl. Acad. Sci. USA 2007, 104, 15988–15993. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Wennekers, T. From Structure to Activity: Using Centrality Measures to Predict Neuronal Activity. Int. J. Neural Syst. 2018, 28, 1750013. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.M. Wound Repair. In Inflammation: Basic Principles and Clinical Correlates; Gallin, J.I., Goldstein, I.M., Snyderman, R., Eds.; Raven Press: New York, NY, USA, 1992; pp. 806–819. [Google Scholar]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, I.B.M.; Pidaparti, R. Influence of Pathogens and Mechanical Stimuli in Inflammation. Bioengineering 2019, 6, 55. https://doi.org/10.3390/bioengineering6020055
Ibrahim IBM, Pidaparti R. Influence of Pathogens and Mechanical Stimuli in Inflammation. Bioengineering. 2019; 6(2):55. https://doi.org/10.3390/bioengineering6020055
Chicago/Turabian StyleIbrahim, Israr B.M., and Ramana Pidaparti. 2019. "Influence of Pathogens and Mechanical Stimuli in Inflammation" Bioengineering 6, no. 2: 55. https://doi.org/10.3390/bioengineering6020055
APA StyleIbrahim, I. B. M., & Pidaparti, R. (2019). Influence of Pathogens and Mechanical Stimuli in Inflammation. Bioengineering, 6(2), 55. https://doi.org/10.3390/bioengineering6020055