
Effects of Heterologous tRNA Modifications on the Production of Proteins Containing Noncanonical Amino Acids


Abstract
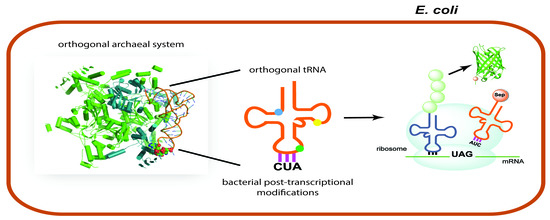
{kind=link}
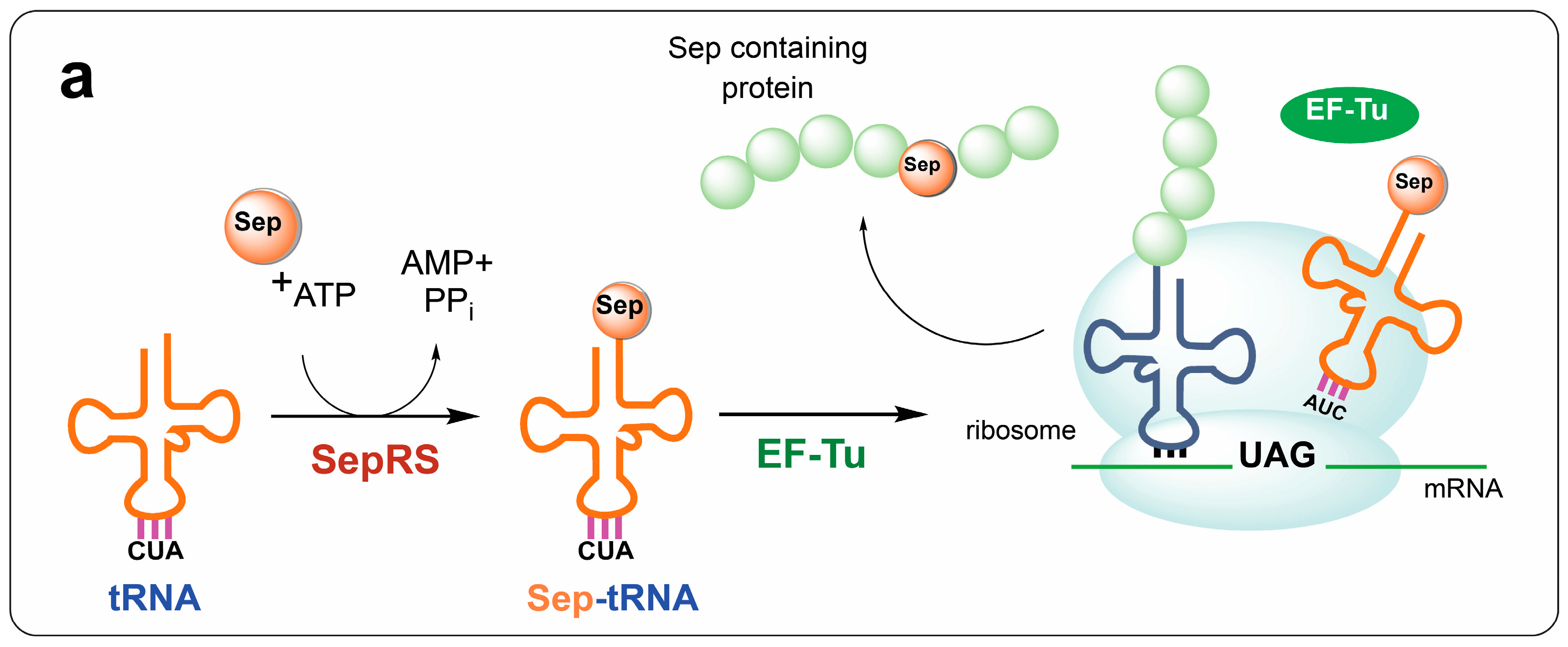{kind=link}
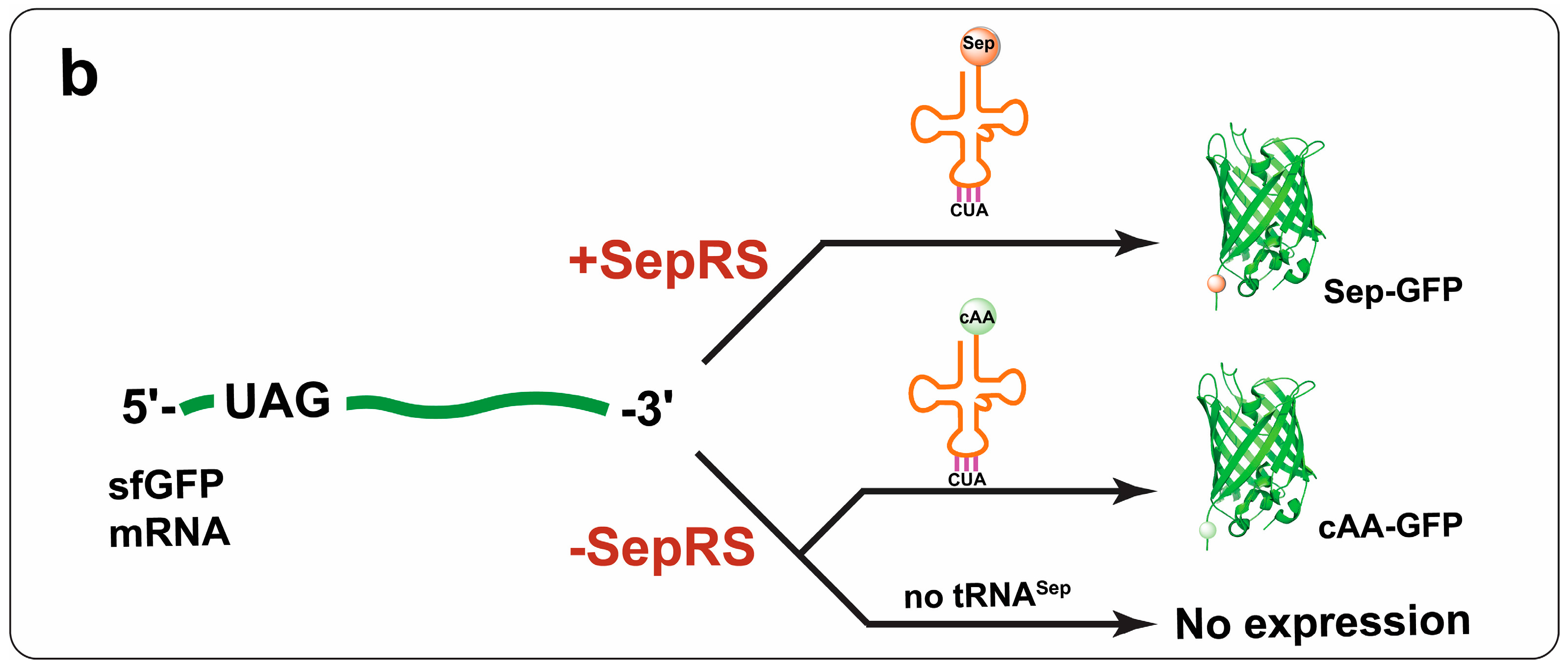{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Plasmid Constructions
2.2. Growth Media
2.3. SfGFP-Based Activity Assays
2.4. SfGFP Purification and Phos-Tag Analysis
3. Results and Discussion
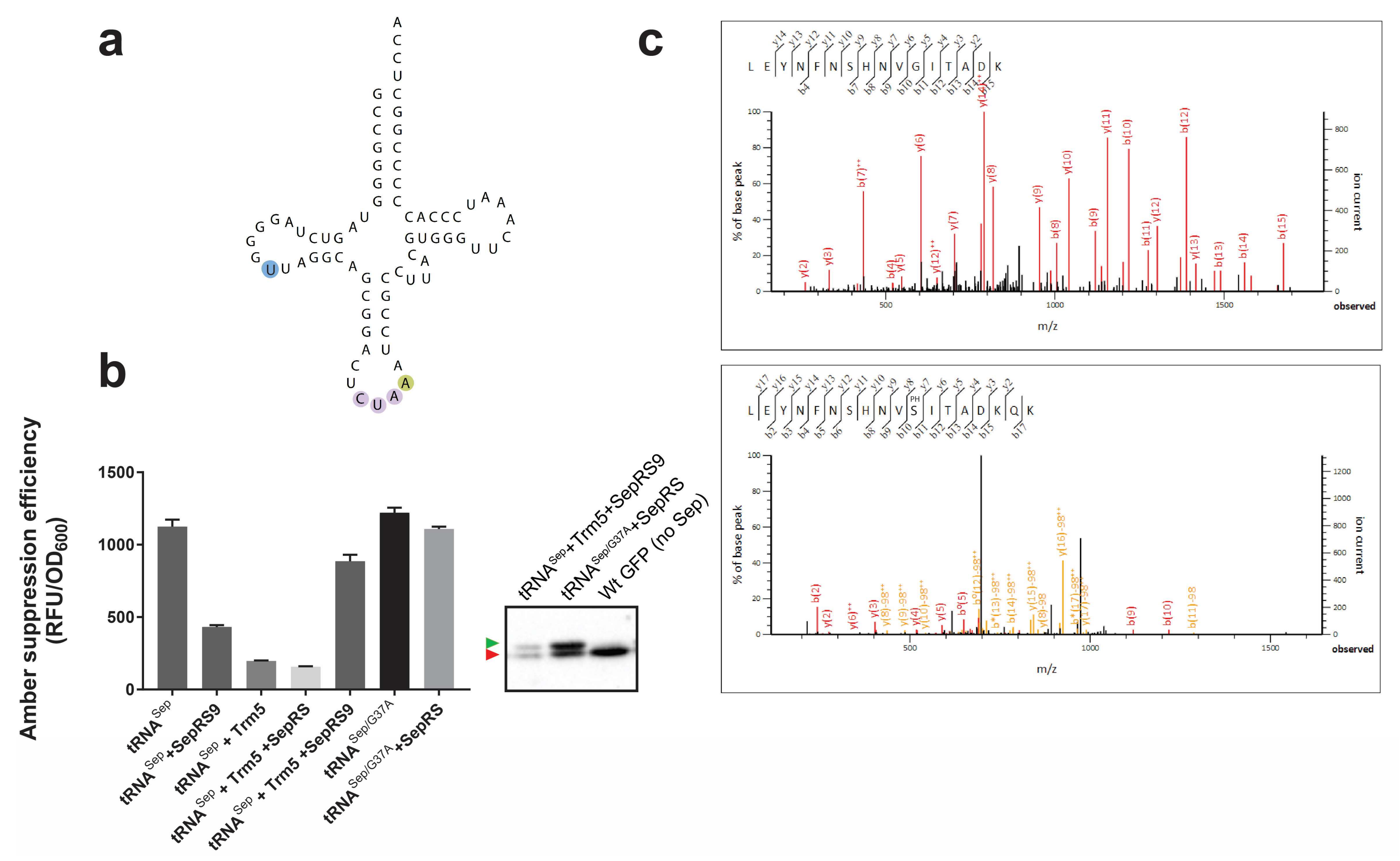
3.1. Implications of Post-Transcriptional Modifications in Orthogonal tRNAs
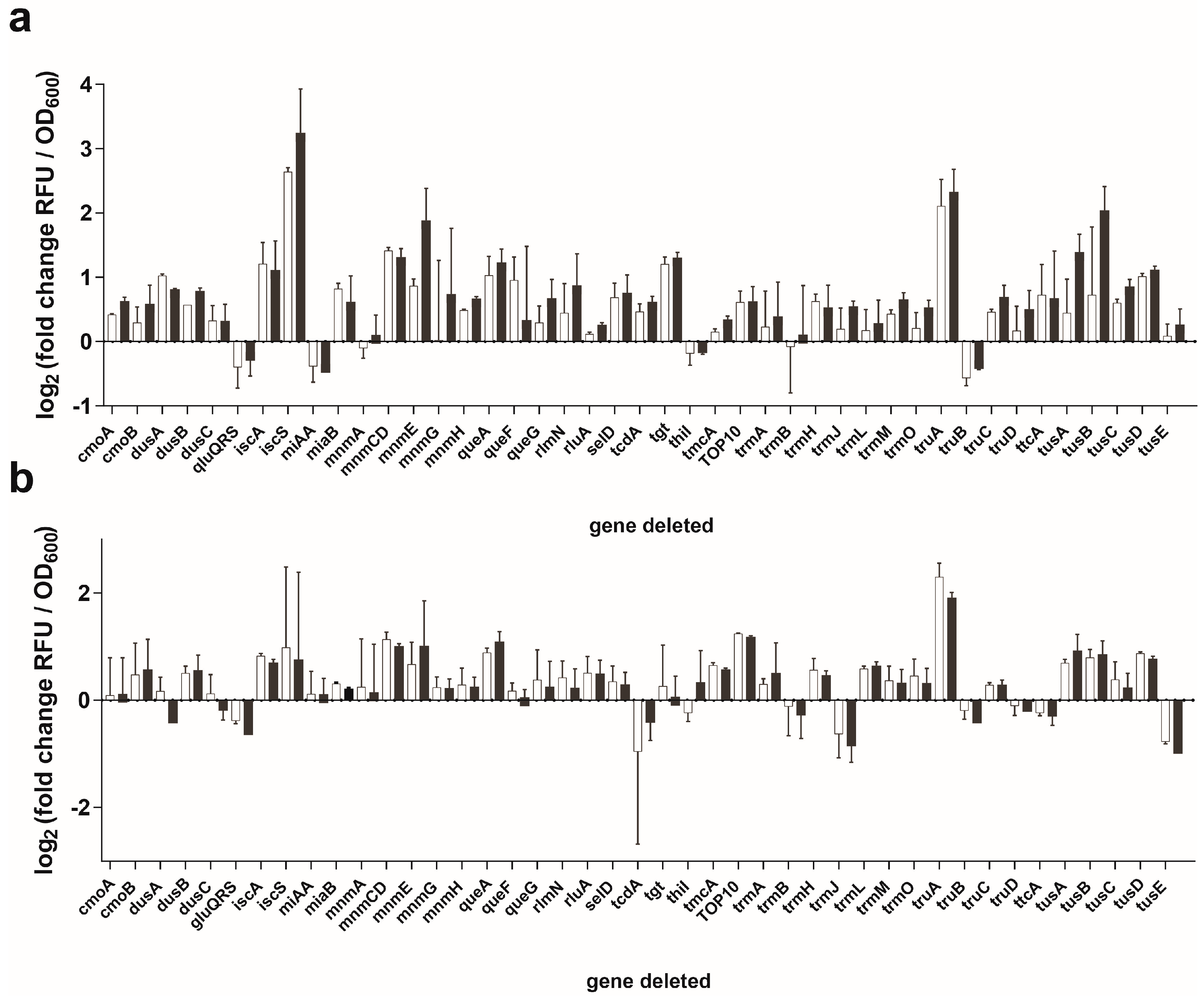
3.2. Sep-OTS Activity in the Absence of Individual Post-transcriptional Modification Enzymes
3.3. Effects of Post-transcriptional Modifications on the Orthogonality of tRNASep/G37A
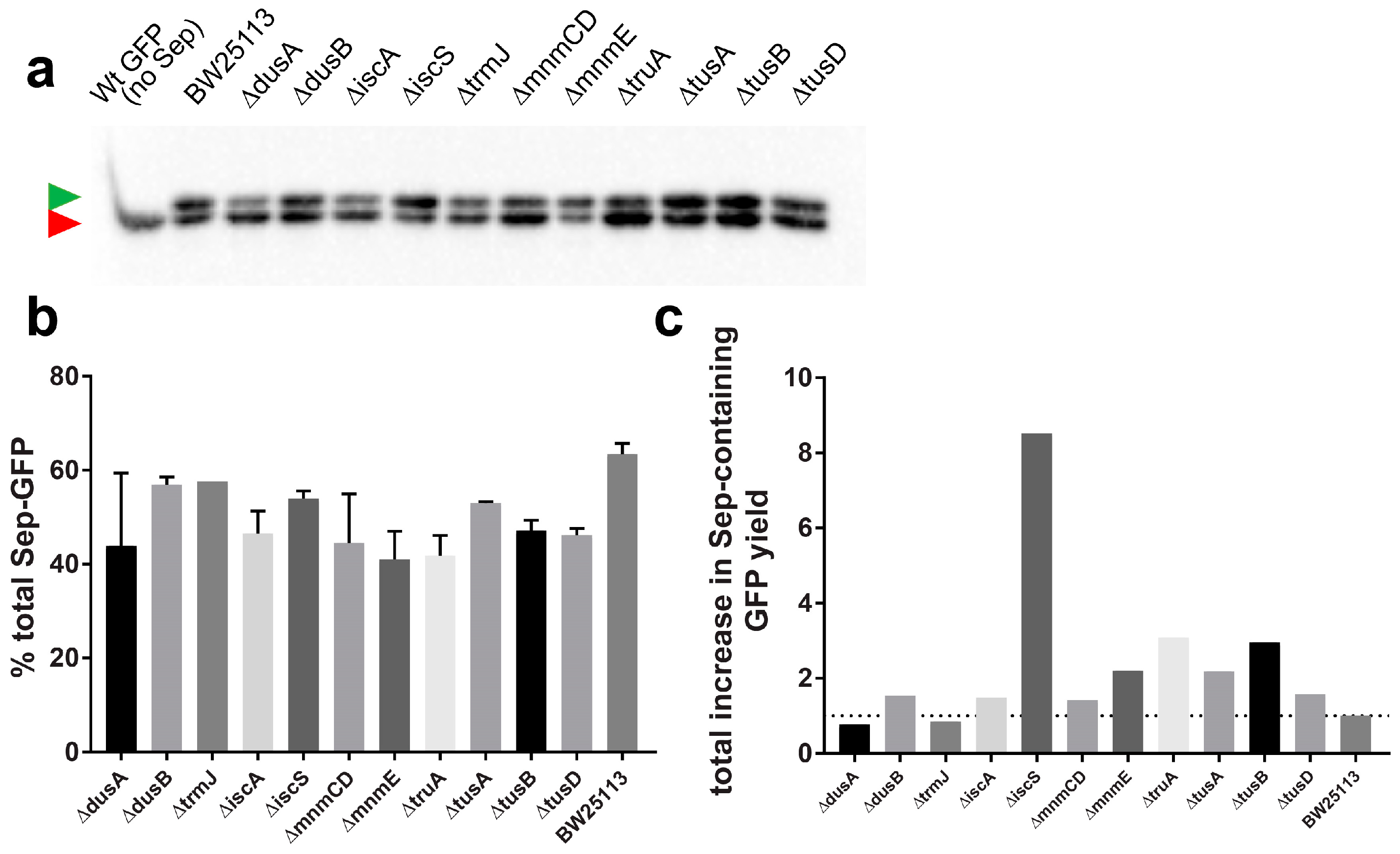
3.4. Deletion of iscS Leads to a Significant Increase of Sep-Containing sfGFP
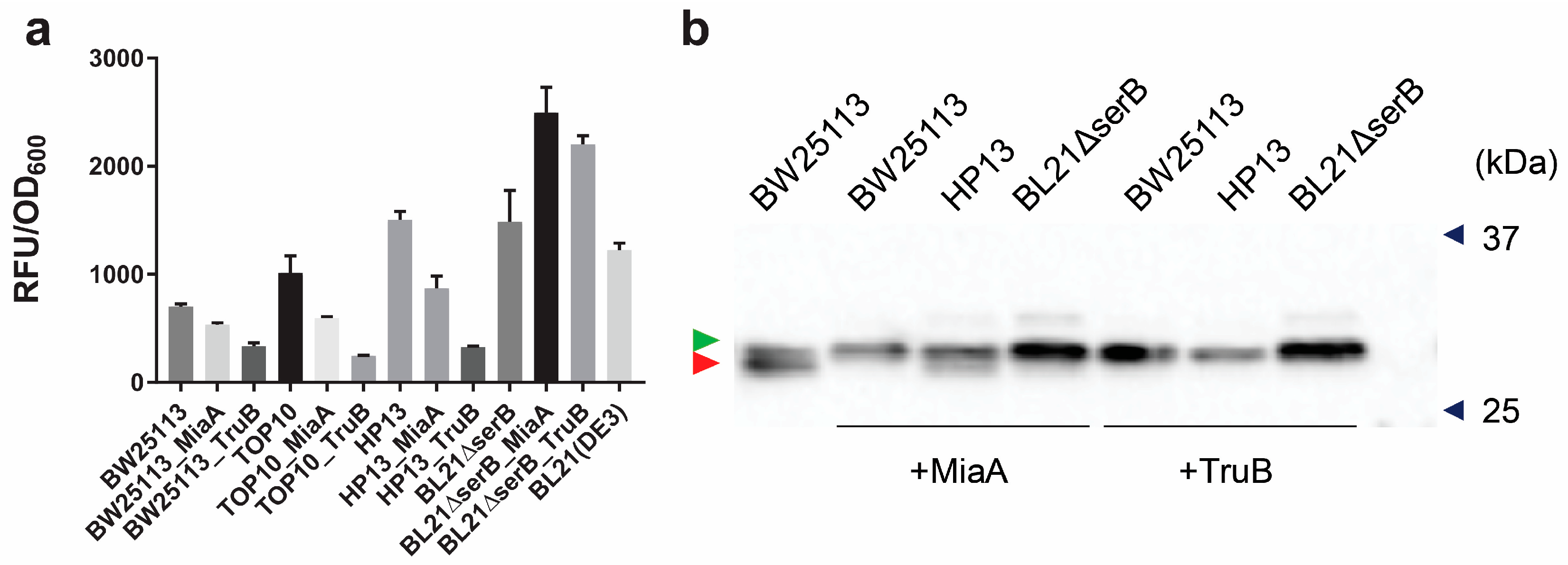
3.5. Overexpression of MiaA and TruB Leads to a Higher Purity of Sep-Containing sfGFP
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gagner, J.E.; Kim, W.; Chaikof, E.L. Designing protein-based biomaterials for medical applications. Acta Biomater. 2014, 10, 1542–1557. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Lercher, L.; Spicer, C.D.; Davis, B.G. Designing logical codon reassignment—Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Koole, C.; Reynolds, C.A.; Mobarec, J.C.; Hick, C.; Sexton, P.M.; Sakmar, T.P. Genetically encoded photocross-linkers determine the biological binding site of exendin-4 peptide in the N-terminal domain of the intact human glucagon-like peptide-1 receptor (GLP-1R). J. Biol. Chem. 2017, 292, 7131–7144. [Google Scholar] [CrossRef] [PubMed]
- Voller, J.S.; Dulic, M.; Gerling-Driessen, U.I.; Biava, H.; Baumann, T.; Budisa, N.; Gruic-Sovulj, I.; Koksch, B. Discovery and Investigation of Natural Editing Function against Artificial Amino Acids in Protein Translation. ACS Cent. Sci. 2017, 3, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Tirrell, D.A. Biosynthesis of a highly stable coiled-coil protein containing hexafluoroleucine in an engineered bacterial host. J. Am. Chem. Soc. 2001, 123, 11089–11090. [Google Scholar] [CrossRef] [PubMed]
- Melo Czekster, C.; Robertson, W.E.; Walker, A.S.; Söll, D.; Schepartz, A. In Vivo Biosynthesis of a beta-Amino Acid-Containing Protein. J. Am. Chem. Soc. 2016, 138, 5194–5197. [Google Scholar] [CrossRef] [PubMed]
- Carrico, I.S.; Maskarinec, S.A.; Heilshorn, S.C.; Mock, M.L.; Liu, J.C.; Nowatzki, P.J.; Franck, C.; Ravichandran, G.; Tirrell, D.A. Lithographic patterning of photoreactive cell-adhesive proteins. J. Am. Chem. Soc. 2007, 129, 4874–4875. [Google Scholar] [CrossRef] [PubMed]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Moglich, A.; et al. Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017, 18, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Ha, S.; Ahn, J.; Kim, R.; Kim, S.; Lee, Y.; Kim, J.; Söll, D.; Lee, H.Y.; Park, H.S. A chemical biology route to site-specific authentic protein modifications. Science 2016, 354, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.M.; Vargas-Rodriguez, O.; Söll, D.; Crnkovic, A. The central role of tRNA in genetic code expansion. Biochim. Biophys. Acta 2017, 1861, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yu, A.C.; Chan, T.F. Efforts and Challenges in Engineering the Genetic Code. Life 2017, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Prat, L.; Aerni, H.R.; Ling, J.; Merryman, C.; Glass, J.I.; Rinehart, J.; Söll, D. Transfer RNA misidentification scrambles sense codon recoding. Chembiochem 2013, 14, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hohn, M.J.; Umehara, T.; Guo, L.T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Söll, D. Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.C.; Yim, A.K.; Mat, W.K.; Tong, A.H.; Lok, S.; Xue, H.; Tsui, S.K.; Wong, J.T.; Chan, T.F. Mutations enabling displacement of tryptophan by 4-fluorotryptophan as a canonical amino acid of the genetic code. Genome Biol. Evol. 2014, 6, 629–641. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol. Syst. Biol. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Wang, Y.S.; Nakamura, A.; Guo, L.T.; Söll, D.; Umehara, T. Pyrrolysyl-tRNA synthetase variants reveal ancestral aminoacylation function. FEBS Lett. 2013, 587, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Englert, M.; Vargas-Rodriguez, O.; Reynolds, N.M.; Wang, Y.S.; Söll, D.; Umehara, T. A genomically modified Escherichia coli strain carrying an orthogonal E. coli histidyl-tRNA synthetase•tRNAHis pair. Biochim. Biophys. Acta 2017, 1861, 3009–3015. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Väre, V.Y.; Eruysal, E.R.; Narendran, A.; Sarachan, K.L.; Agris, P.F. Chemical and Conformational Diversity of Modified Nucleosides Affects tRNA Structure and Function. Biomolecules 2017, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA modifications: Impact on structure and thermal adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Alfonzo, J.D. Transfer RNA modifications: Nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 2013, 4, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Giege, R.; Sissler, M.; Florentz, C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998, 26, 5017–5035. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; Westhof, E. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016, 44, 8020–8040. [Google Scholar] [CrossRef] [PubMed]
- Christian, T.; Hou, Y.M. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol. 2007, 373, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Shimada, M.; Armache, A.; Li, C.H.; Miller, R.M.; Lin, S.; Yang, A.; Dill, B.D.; Molina, H.; Park, H.S.; et al. Chromatin Kinases Act on Transcription Factors and Histone Tails in Regulation of Inducible Transcription. Mol. Cell 2016, 64, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Pirman, N.L.; Barber, K.W.; Aerni, H.R.; Ma, N.J.; Haimovich, A.D.; Rogulina, S.; Isaacs, F.J.; Rinehart, J. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat. Commun. 2015, 6, 8130. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, D.T.; Sachdeva, A.; Wang, K.; Haq, T.; Kazlauskaite, A.; Hancock, S.M.; Huguenin-Dezot, N.; Muqit, M.M.; Fry, A.M.; Bayliss, R.; et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat. Chem. Biol. 2015, 11, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Oh, S.; Yang, A.; Kim, J.; Söll, D.; Lee, D.; Park, H.S. A Facile Strategy for Selective Incorporation of Phosphoserine into Histones. Angew. Chem. Int. Ed. 2013, 52, 5771–5775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.M.; Liu, C.; Slater, S.; Hou, Y.M. Aminoacylation of tRNA with phosphoserine for synthesis of cysteinyl-tRNACys. Nat. Struct. Mol. Biol. 2008, 15, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Cotlet, M.; Goodwin, P.M.; Waldo, G.S.; Werner, J.H. A comparison of the fluorescence dynamics of single molecules of a green fluorescent protein: One- versus two-photon excitation. Chemphyschem 2006, 7, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hohn, M.J.; Park, H.S.; O’Donoghue, P.; Schnitzbauer, M.; Söll, D. Emergence of the universal genetic code imprinted in an RNA record. Proc. Natl. Acad. Sci. USA 2006, 103, 18095–18100. [Google Scholar] [CrossRef] [PubMed]
- Gefter, M.L.; Russell, R.L. Role modifications in tyrosine transfer RNA: A modified base affecting ribosome binding. J. Mol. Biol. 1969, 39, 145–157. [Google Scholar] [CrossRef]
- Nameki, N.; Tamura, K.; Asahara, H.; Hasegawa, T. Recognition of tRNAGly by three widely diverged glycyl-tRNA synthetases. J. Mol. Biol. 1997, 268, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Biddle, W.; Schmitt, M.A.; Fisk, J.D. Modification of orthogonal tRNAs: Unexpected consequences for sense codon reassignment. Nucleic Acids Res. 2016, 44, 10042–10050. [Google Scholar] [CrossRef] [PubMed]
- Bon Ramos, A.; Bao, L.; Turner, B.; de Crecy-Lagard, V.; Iwata-Reuyl, D. QueF-Like, a Non-Homologous Archaeosine Synthase from the Crenarchaeota. Biomolecules 2017, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Tramontano, A.; Cavallo, L. Mg2+ binding and archaeosine modification stabilize the G15–C48 Levitt base pair in tRNAs. RNA 2007, 13, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Waller, J.C.; Ellens, K.W.; Hasnain, G.; Alvarez, S.; Rocca, J.R.; Hanson, A.D. Evidence that the Folate-Dependent Proteins YgfZ and MnmEG Have Opposing Effects on Growth and on Activity of the Iron-Sulfur Enzyme MiaB. J. Bacteriol. 2012, 194, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Arifuzzaman, M.; Maeda, M.; Itoh, A.; Nishikata, K.; Takita, C.; Saito, R.; Ara, T.; Nakahigashi, K.; Huang, H.C.; Hirai, A.; et al. Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 2006, 16, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Alfonzo, J.D. Posttranscriptional RNA Modifications: Playing metabolic games in a cell’s chemical Legoland. Chem. Biol. 2014, 21, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, P.C.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t6A), a universal modification of tRNA. RNA Biol. 2014, 11, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Cavuzic, M.; Liu, Y. Biosynthesis of Sulfur-Containing tRNA Modifications: A Comparison of Bacterial, Archaeal, and Eukaryotic Pathways. Biomolecules 2017, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, J.B.; Aerni, H.R.; Rogulina, S.; Liu, Y.; Rinehart, J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem. Biol. 2014, 9, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Hayashi, A.; Iraha, F.; Sato, A.; Ohtake, K.; Yokoyama, S.; Sakamoto, K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010, 38, 8188–8195. [Google Scholar] [CrossRef] [PubMed]
- Kosako, H. Phos-tag Western blotting for detecting stoichiometric protein phosphorylation in cells. Nature 2009. [Google Scholar] [CrossRef]
- Laten, H.; Gorman, J.; Bock, R.M. Isopentenyladenosine deficient tRNA from an antisuppressor mutant of Saccharomyces cerevisiae. Nucleic Acids Res. 1978, 5, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, T.; Poulter, C.D. Escherichia coli dimethylallyl diphosphate: tRNA dimethylallyltransferase: Essential elements for recognition of tRNA substrates within the anticodon stem-loop. Biochemistry 2000, 39, 6546–6553. [Google Scholar] [CrossRef] [PubMed]
- Keffer-Wilkes, L.C.; Veerareddygari, G.R.; Kothe, U. RNA modification enzyme TruB is a tRNA chaperone. Proc. Natl. Acad. Sci. USA 2016, 113, 14306–14311. [Google Scholar] [CrossRef] [PubMed]
- Waegeman, H.; Beauprez, J.; Moens, H.; Maertens, J.; De Mey, M.; Foulquie-Moreno, M.R.; Heijnen, J.J.; Charlier, D.; Soetaert, W. Effect of iclR and arcA knockouts on biomass formation and metabolic fluxes in Escherichia coli K12 and its implications on understanding the metabolism of Escherichia coli BL21 (DE3). BMC Microbiol. 2011, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.M.; Reynolds, N.M.; Rivera, K.; Connolly, M.; Guo, L.T.; Ling, J.; Pappin, D.J.; Church, G.M.; Söll, D. Efficient Reassignment of a Frequent Serine Codon in Wild-Type Escherichia coli. ACS Synth. Biol. 2016, 5, 163–171. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crnković, A.; Vargas-Rodriguez, O.; Merkuryev, A.; Söll, D. Effects of Heterologous tRNA Modifications on the Production of Proteins Containing Noncanonical Amino Acids. Bioengineering 2018, 5, 11. https://doi.org/10.3390/bioengineering5010011
Crnković A, Vargas-Rodriguez O, Merkuryev A, Söll D. Effects of Heterologous tRNA Modifications on the Production of Proteins Containing Noncanonical Amino Acids. Bioengineering. 2018; 5(1):11. https://doi.org/10.3390/bioengineering5010011
Chicago/Turabian StyleCrnković, Ana, Oscar Vargas-Rodriguez, Anna Merkuryev, and Dieter Söll. 2018. "Effects of Heterologous tRNA Modifications on the Production of Proteins Containing Noncanonical Amino Acids" Bioengineering 5, no. 1: 11. https://doi.org/10.3390/bioengineering5010011
APA StyleCrnković, A., Vargas-Rodriguez, O., Merkuryev, A., & Söll, D. (2018). Effects of Heterologous tRNA Modifications on the Production of Proteins Containing Noncanonical Amino Acids. Bioengineering, 5(1), 11. https://doi.org/10.3390/bioengineering5010011