
Hypoxic Three-Dimensional Scaffold-Free Aggregate Cultivation of Mesenchymal Stem Cells in a Stirred Tank Reactor




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
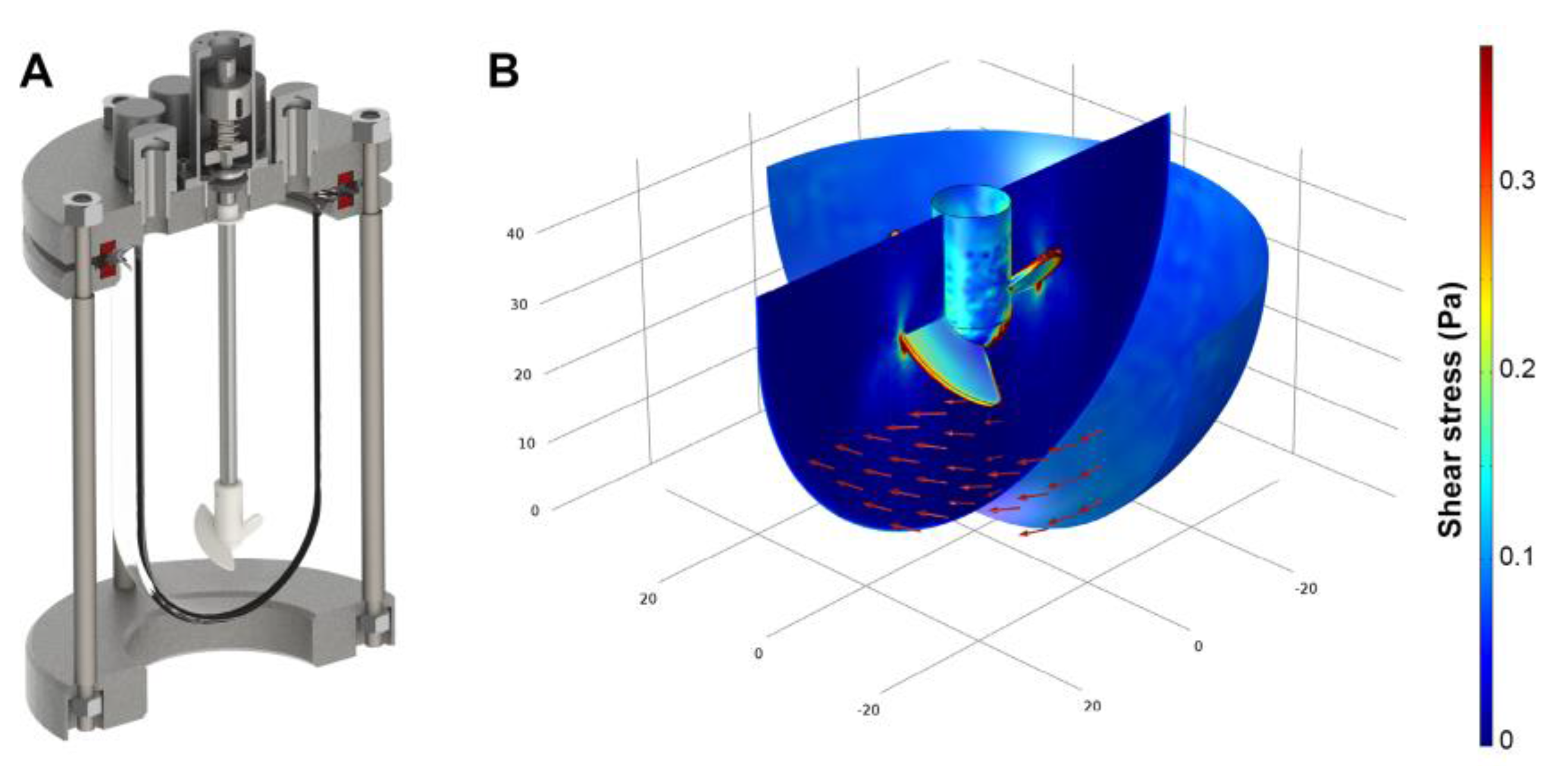
2.1. Bioreactor Design
2.2. Compuitational Fluid Dyamics
2.3. Cell Culture
2.4. Bioreactor Cultivation
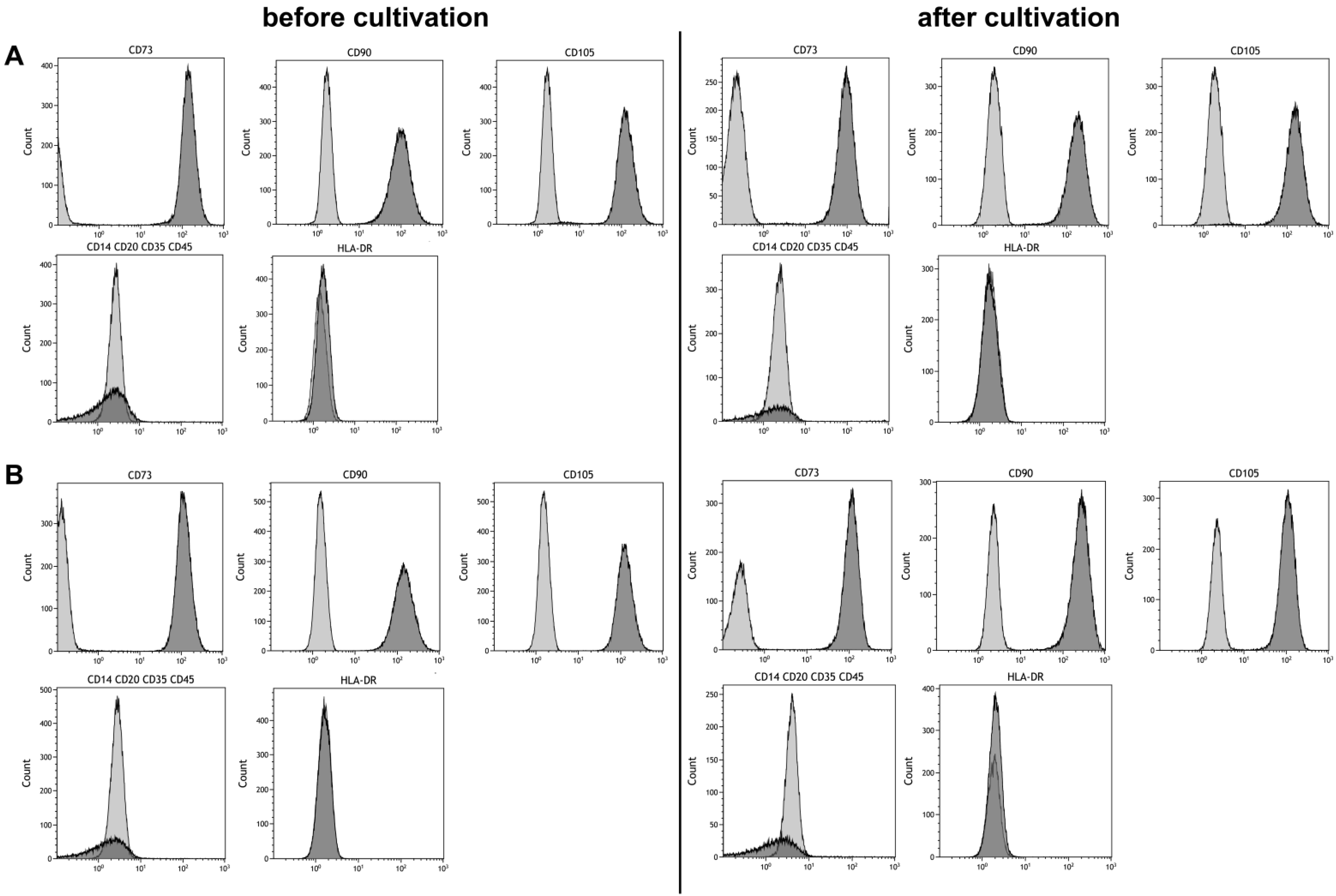
2.5. Phenotyping
2.6. Differentiation
2.7. Histologic Stainings
2.8. Statistical Analysis
3. Results
3.1. Shear Stress Estimation By Computational Fluid Dynamics
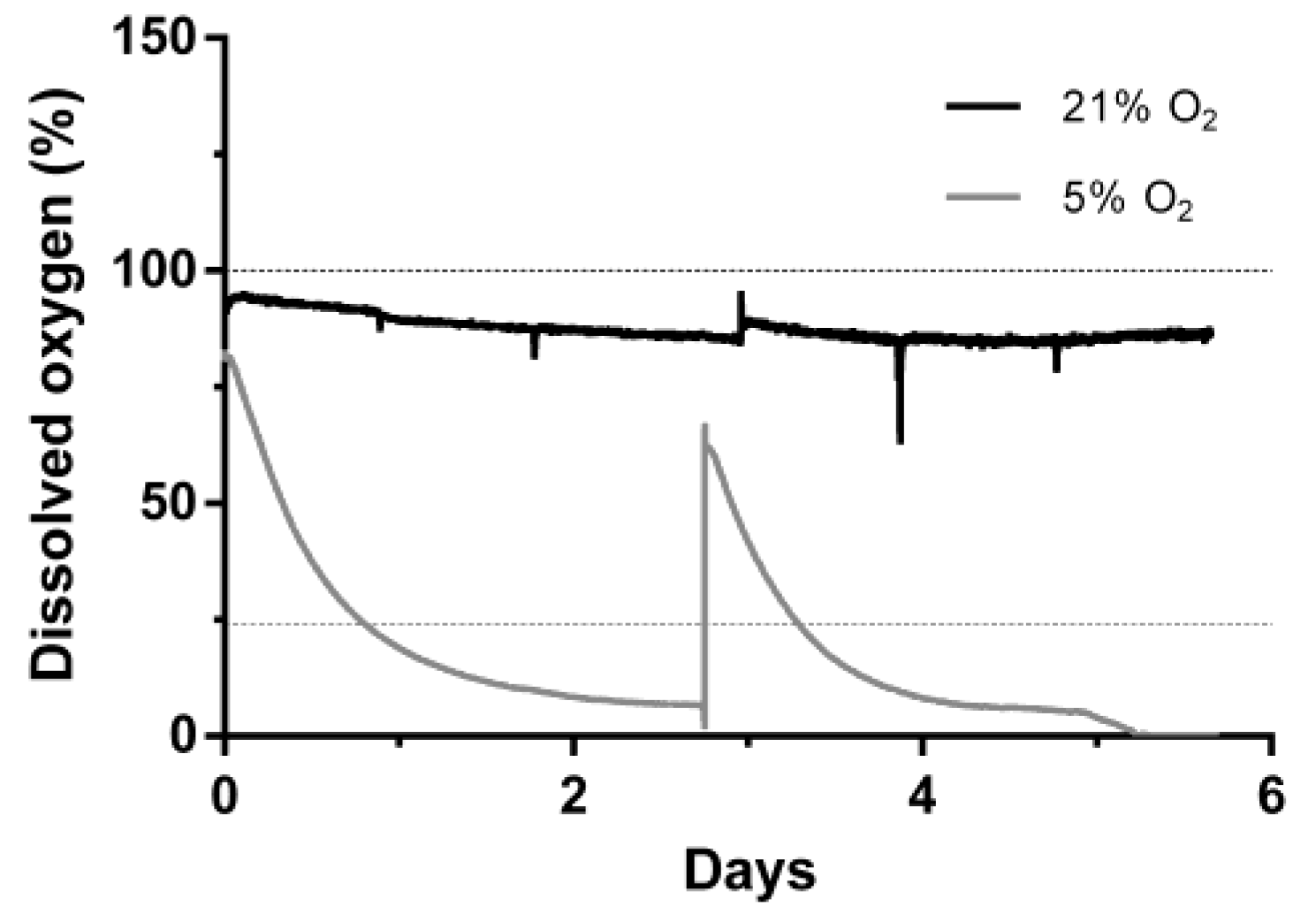
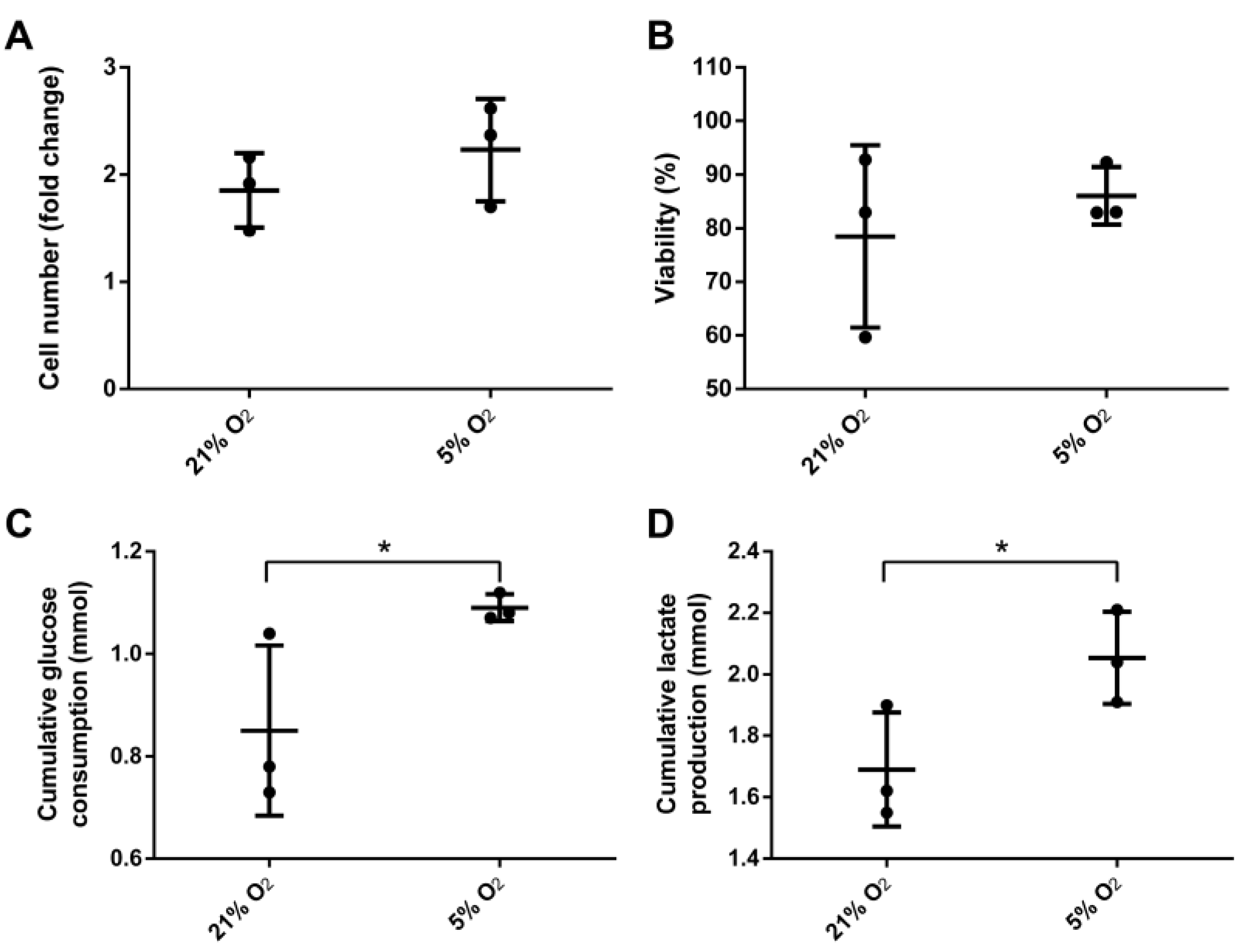
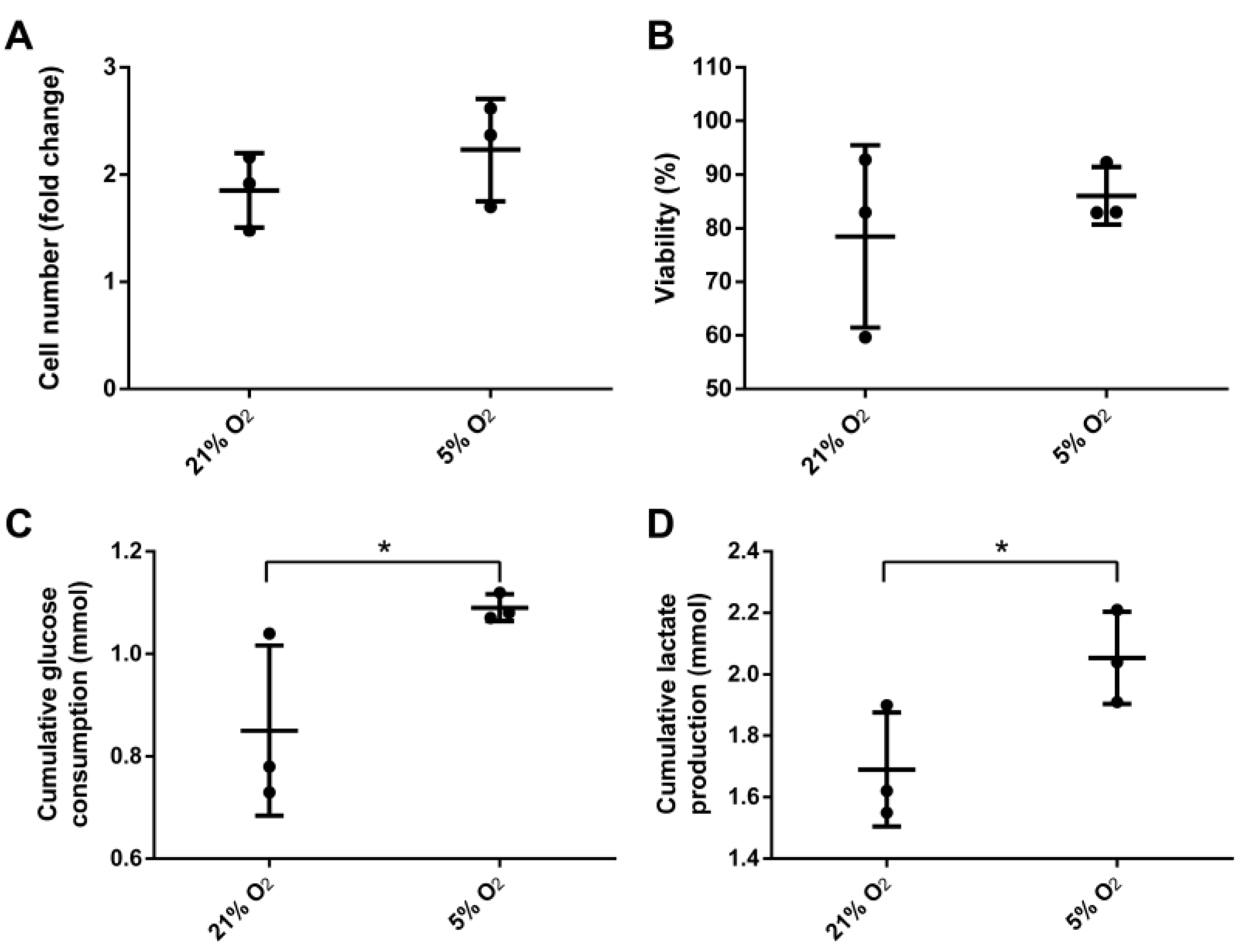
3.2. Bioreactor Cultivation
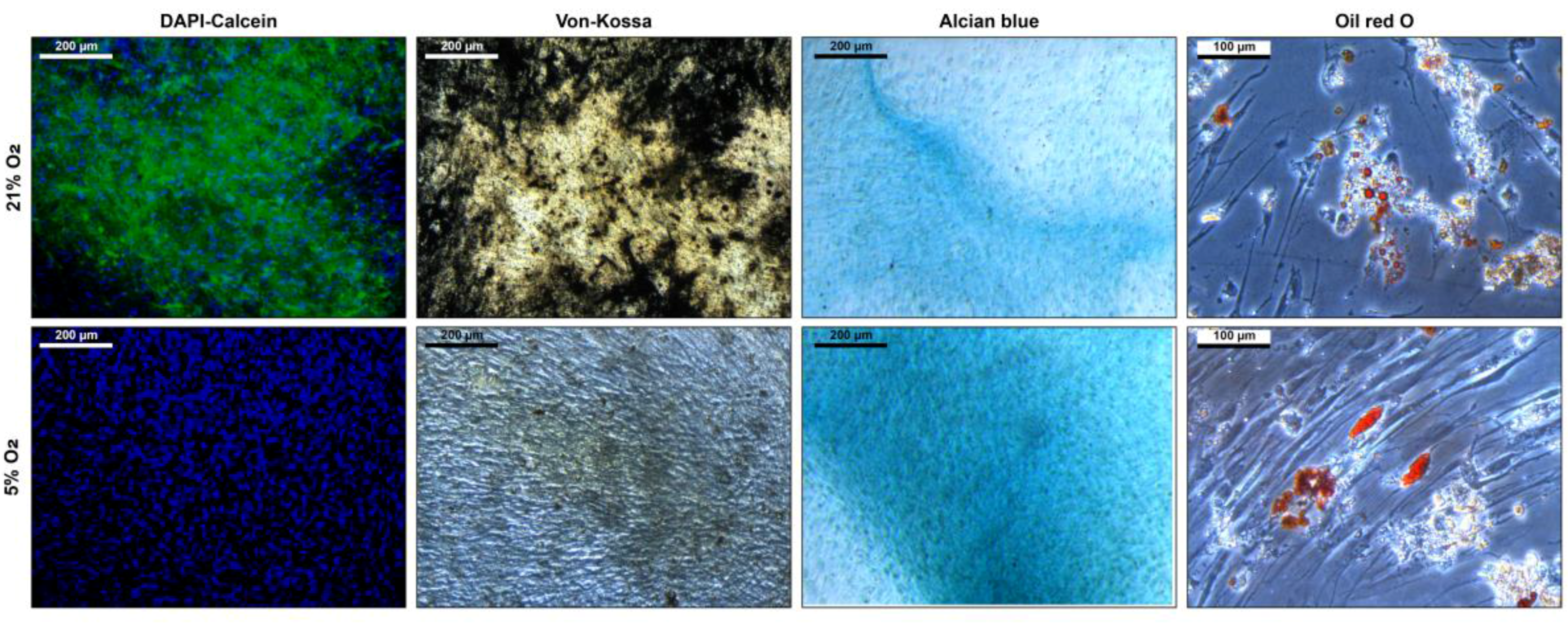
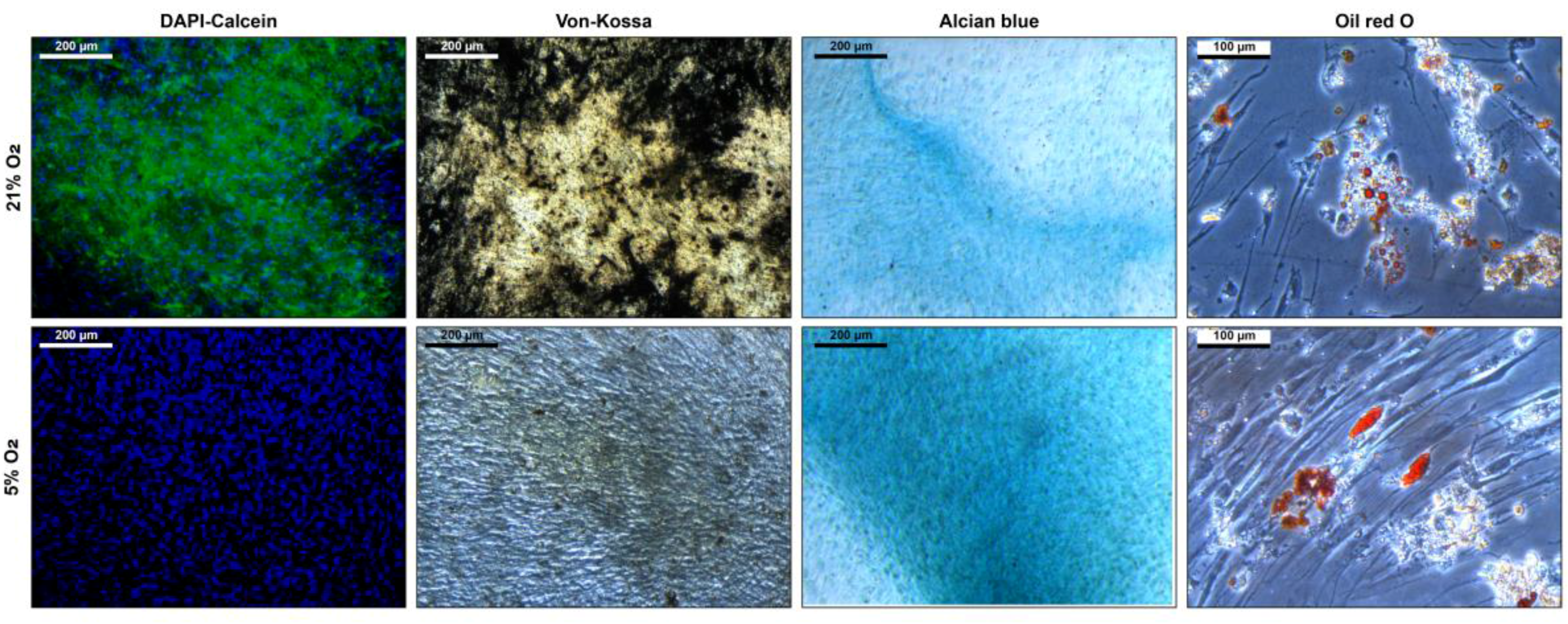
3.3. Stem Cell Properties
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Kim, M.K.; Shin, M.S.; Lee, H.J.; Ko, J.H.; Wee, W.R.; Lee, J.H. The Anti-Inflammatory and Anti-Angiogenic Role of Mesenchymal Stem Cells in Corneal Wound Healing Following Chemical Injury. Stem Cells 2008, 26, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Capla, A.I. Adult Mesenchymal Stem Cells for Tissue Engineering Versus Regenerative Medicine. J. Cell. Physiol. 2007, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.G.; Carvalho, M.M.; Sousa, N.; Salgado, A.J. Mesenchymal stem cells secretome: A new paradigm for central nervous system regeneration? Cell. Mol. Life Sci. 2013, 70, 3871–3882. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, S.H.; Levy, O.; Inamdar, M.S.; Karp, J.M. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell. Stem Cell. 2012, 10, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J. The mesenchymal stromal cells dilemma-does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Von Bahr, L.; Sundberg, B.; Lonnies, L.; Sander, B.; Karbach, H.; Hagglund, H.; Ljungman, P.; Gustafsson, B.; Karlsson, H.; Le Blanc, K.; et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol. Blood Marrow Transplant. 2012, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; McIntosh, K.; Zvonic, S.; Garrett, S.; Floyd, Z.E.; Kloster, A.; Di Halvorsen, Y.; Storms, R.W.; Goh, B.; Kilroy, G.; et al. Immunophenotype of Human Adipose-Derived Cells: Temporal Changes in Stromal-Associated and Stem Cell-Associated Markers. Stem Cells 2006, 24, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Bartosh, T.J.; Ylostalo, J.H.; Mohammadipoor, A.; Bazhanov, N.; Coble, K.; Claypool, K.; Lee, R.H.; Choi, H.; Prockop, D.J. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl. Acad. Sci. 2010, 107, 13724–13729. [Google Scholar] [CrossRef] [PubMed]
- Frith, J.E.; Thomson, B.; Genever, P.G. Dynamic Three-Dimensional Culture Methods Enhance Mesenchymal Stem Cell Properties and Increase Therapeutic Potential. Tissue Eng. Part. C Methods 2010, 16, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Ylöstalo, J.H.; Bartosh, T.J.; Coble, K.; Prockop, D.J. Human mesenchymal stem/stromal cells cultured as spheroids are self-activated to produce prostaglandin E2 that directs stimulated macrophages into an anti-inflammatory phenotype. Stem Cells 2012, 30, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.-C.; Chen, S.-Y.; Li, J.-R.; Young, T.-H. Short-Term Spheroid Formation Enhances the Regenerative Capacity of Adipose-Derived Stem Cells by Promoting Stemness, Angiogenesis, and Chemotaxis. Stem Cells Transl. Med. 2013, 2, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, C.; Büth, H.; Thielecke, H. A scaffold-free in vitro model for osteogenesis of human mesenchymal stem cells. Tissue Cell 2011, 43, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Pérez, J.; Ballesteros, P.; Cerdán, S. Microscopic images of intraspheroidal pH by 1H magnetic resonance chemical shift imaging of pH sensitive indicators. Magn. Reson. Mater. Physics, Biol. Med. 2005, 18, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Bhang, S.H.; Cho, S.W.; La, W.G.; Lee, T.J.; Yang, H.S.; Sun, A.Y.; Baek, S.H.; Rhie, J.W.; Kim, B.S. Angiogenesis in ischemic tissue produced by spheroid grafting of human adipose-derived stromal cells. Biomaterials 2011, 32, 2734–2747. [Google Scholar] [CrossRef] [PubMed]
- Alimperti, S.; Lei, P.; Wen, Y.; Tian, J.; Campbell, A.M.; Andreadis, S.T. Serum-free spheroid suspension culture maintains mesenchymal stem cell proliferation and differentiation potential. Biotechnol. Prog. 2014, 30, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, P.R.; McDevitt, T.C. Scaffold-free culture of mesenchymal stem cell spheroids in suspension preserves multilineage potential. Cell. Tissue Res. 2012, 347, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S.; Rameshwar, P.; Chang, V.; Bandar, P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 2002, 125, 1679–1682. [Google Scholar] [CrossRef]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO2 Distributions in the Bone Marrow Hematopoietic Compartment. II. Modified Kroghian Models. Biophys. J. 2001, 81, 685–696. [Google Scholar] [CrossRef]
- Lavrentieva, A.; Majore, I.; Kasper, C.; Hass, R. Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell. Commun. Signal. 2010, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Buravkova, L.B.; Andreeva, E.R.; Gogvadze, V.; Zhivotovsky, B. Mesenchymal stem cells and hypoxia: Where are we? Mitochondrion 2014, 19, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Takeda-Kawaguchi, T.; Tezuka, Y.; Kunisada, T.; Shibata, T.; Tezuka, K.I. Hypoxia enhances colony formation and proliferation but inhibits differentiation of human dental pulp cells. Arch. Oral Biol. 2010, 55, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Grayson, W.L.; Zhao, F.; Bunnell, B.; Ma, T. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2007, 358, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Chen, Y.J.; Yew, T.L.; Chen, L.L.; Wang, J.Y.; Chiu, C.H.; Hung, S.C. Hypoxia inhibits senescence and maintains mesenchymal stem cell properties through down-regulation of E2A-p21 by HIF-TWIST. Blood 2011, 117, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Estrada, J.C.; Albo, C.; Benguría, A.; Dopazo, A.; López-Romero, P.; Carrera-Quintanar, L.; Roche, E.; Clemente, E.P.; Enríquez, J.A.; Bernad, A.; et al. Culture of human mesenchymal stem cells at low oxygen tension improves growth and genetic stability by activating glycolysis. Cell. Death Differ. 2012, 19, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Nold, P.; Hackstein, H.; Riedlinger, T.; Kasper, C.; Neumann, A.; Mernberger, M.; Fölsch, C.; Schmitt, J.; Fuchs-Winkelmann, S.; Barckhausen, C.; et al. Immunosuppressive capabilities of mesenchymal stromal cells are maintained under hypoxic growth conditions and after gamma irradiation. Cytotherapy 2015, 17, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, F.; Andrade, P.Z.; Boura, J.S.; Abecasis, M.M.; Da Silva, C.L.; Cabral, J.M.S. Ex vivo expansion of human mesenchymal stem cells: A more effective cell proliferation kinetics and metabolism under hypoxia. J. Cell. Physiol. 2010, 223, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, F.; Berr, K.; Naujoks, C.; Hassel, A.; Hentschel, M.; Depprich, R.; Kubler, N.R.; Meyer, U.; Wiesmann, H.-P.; Kögler, G.; et al. Generation and differentiation of microtissues from multipotent precursor cells for use in tissue engineering. Nat. Protoc. 2011, 6, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Tsai, A.-C.; Li, Y.; Ma, T. Three-Dimensional Aggregates of Mesenchymal Stem Cells: Cellular Mechanisms, Biological Properties, and Applications. Tissue Eng. Part. B Rev. 2014, 20, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Kleinhans, C.; Mohan, R.R.; Vacun, G.; Schwarz, T.; Haller, B.; Sun, Y.; Kahlig, A.; Kluger, P.; Finne-Wistrand, A.; Walles, H.; et al. A perfusion bioreactor system efficiently generates cell-loaded bone substitute materials for addressing critical size bone defects. Biotechnol. J. 2015, 10, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Spitz, S.; Fischer, M.; Handschuh, S.; Glösmann, M.; Friemert, B.; Egerbacher, M.; Kasper, C. Application of a Parallelizable Perfusion Bioreactor for Physiologic 3D Cell Culture. Cells Tissues Organs 2017, 203, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, P.R.; Cooke, M.T.; Saeed, R.; Kinney, M.A.; Fridley, K.M.; McDevitt, T.C. Stiffening of human mesenchymal stem cell spheroid microenvironments induced by incorporation of gelatin microparticles. J. Mech. Behav. Biomed. Mater. 2012, 11, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Malladi, P.; Xu, Y.; Chiou, M.; Giaccia, A.J.; Michael, T.; Longaker, M.T. Effect of reduced oxygen tension on chondrogenesis and osteogenesis in adipose-derived mesenchymal cells. AJP Cell Physiol. 2006, 290, C1139–C1146. [Google Scholar] [CrossRef] [PubMed]
- Valorani, M.G.; Montelatici, E.; Germani, A.; Biddle, A.; D’Alessandro, D.; Strollo, R.; Patrizi, M.P.; Lazzari, L.; Nye, E.; Otto, W.R.; et al. Pre-culturing human adipose tissue mesenchymal stem cells under hypoxia increases their adipogenic and osteogenic differentiation potentials. Cell. Prolif. 2012, 45, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Hanley, P.J.; Mei, Z.; Durett, A.G.; da Graca Cabreira-Harrison, M.; Klis, M.; Li, W.; Zhao, Y.; Yang, B.; Parsha, K.; Mir, O.; et al. Efficient manufacturing of therapeutic mesenchymal stromal cells with the use of the Quantum Cell Expansion System. Cytotherapy 2014, 16, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Eibes, G.; dos Santos, F.; Andrade, P.Z.; Boura, J.S.; Abecasis, M.M.A.; da Silva, C.L.; Cabral, J.M.S. Maximizing the ex vivo expansion of human mesenchymal stem cells using a microcarrier-based stirred culture system. J. Biotechnol. 2010, 146, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, F.; Campbell, A.; Fernandes-Platzgummer, A.; Andrade, P.Z.; Gimble, J.M.; Wen, Y.; Boucher, S.; Vemuri, M.C.; Da Silva, C.L.; Cabral, J.M.S. A xenogeneic-free bioreactor system for the clinical-scale expansion of human mesenchymal stem/stromal cells. Biotechnol. Bioeng. 2014, 111, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zang, R.; Yang, S.T.; Li, Y. Stem cell engineering in bioreactors for large-scale bioprocessing. Eng. Life Sci. 2014, 14, 4–15. [Google Scholar] [CrossRef]
- Nienow, A.W.; Rafiq, Q.A.; Coopman, K.; Hewitt, C.J. A potentially scalable method for the harvesting of hMSCs from microcarriers. Biochem. Eng. J. 2014, 85, 79–88. [Google Scholar] [CrossRef]
- Zhang, Q.; Nguyen, A.L.; Shi, S.; Hill, C.; Wilder-Smith, P.; Krasieva, T.B.; Le, A.D. Three-Dimensional Spheroid Culture of Human Gingiva-Derived Mesenchymal Stem Cells Enhances Mitigation of Chemotherapy-Induced Oral Mucositis. Stem Cells Dev. 2012, 21, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ma, T. Autocrine fibroblast growth factor 2-mediated interactions between human mesenchymal stem cells and the extracellular matrix under varying oxygen tension. J. Cell. Biochem 2013, 114, 716–727. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egger, D.; Schwedhelm, I.; Hansmann, J.; Kasper, C. Hypoxic Three-Dimensional Scaffold-Free Aggregate Cultivation of Mesenchymal Stem Cells in a Stirred Tank Reactor. Bioengineering 2017, 4, 47. https://doi.org/10.3390/bioengineering4020047
Egger D, Schwedhelm I, Hansmann J, Kasper C. Hypoxic Three-Dimensional Scaffold-Free Aggregate Cultivation of Mesenchymal Stem Cells in a Stirred Tank Reactor. Bioengineering. 2017; 4(2):47. https://doi.org/10.3390/bioengineering4020047
Chicago/Turabian StyleEgger, Dominik, Ivo Schwedhelm, Jan Hansmann, and Cornelia Kasper. 2017. "Hypoxic Three-Dimensional Scaffold-Free Aggregate Cultivation of Mesenchymal Stem Cells in a Stirred Tank Reactor" Bioengineering 4, no. 2: 47. https://doi.org/10.3390/bioengineering4020047
APA StyleEgger, D., Schwedhelm, I., Hansmann, J., & Kasper, C. (2017). Hypoxic Three-Dimensional Scaffold-Free Aggregate Cultivation of Mesenchymal Stem Cells in a Stirred Tank Reactor. Bioengineering, 4(2), 47. https://doi.org/10.3390/bioengineering4020047