3.3. Virtual Screening and Docking Studies
With the advancement of computational technologies, virtual screening has become an indispensable component of modern pharmaceutical research. This approach facilitates the identification of bioactive compounds by analyzing molecular structures and predicting their interactions with biological targets [
55,
56]. In this study, the e-pharmacophore model designated as ADDN was employed to screen a library of 47,781 natural compounds retrieved from the PubChem database. Prior to pharmacophore screening, the compound set was refined using the Canvas Property Filter to ensure compliance with essential drug-like properties. Compounds with a fitness score of 1.5 or higher were selected for further molecular docking analysis to evaluate their potential interaction with the active site of the NA protein. This step significantly enhanced the efficiency and reliability of the virtual screening pipeline. The initial phase of molecular docking involved Glide/SP (Standard Precision) docking of 1829 compounds, which served to eliminate weak binders. Subsequently, the most promising candidates were subjected to Glide/XP (Extra Precision) docking for a more rigorous evaluation of binding affinity and interaction patterns.
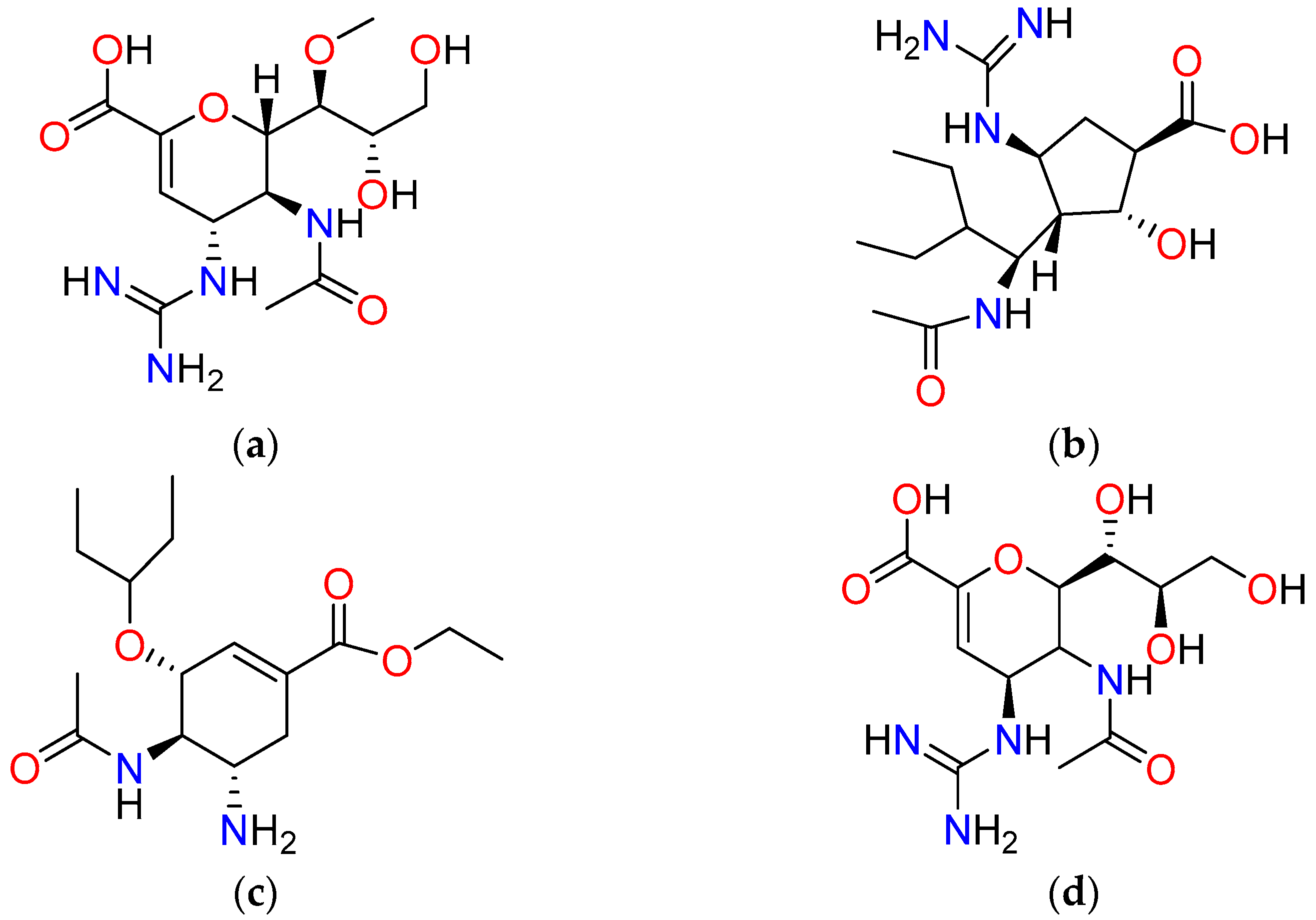
From this two-tiered screening process, three natural compounds emerged as top candidates: CID 102209473 (C1), CID 85692821 (C2), and CID 45379525 (C3). These compounds exhibited the highest docking scores, ranging from –10.503 to –9.433 kcal/mol, as detailed in
Table 2. A comprehensive overview of the three selected compounds—including their synonyms, IUPAC names, natural sources, chemical families, and relevant bibliographic references—is presented in
Table S1.
For comparative purposes, two well-established antiviral agents—Zanamivir, used as the reference molecule, and Oseltamivir—were included in this study due to their proven efficacy against influenza viruses. The XP docking scores were calculated to be −10.168 kcal/mol for Zanamivir and −8.848 kcal/mol for Oseltamivir. Notably, compounds C1 and C2 exhibited XP docking scores lower than both Zanamivir and Oseltamivir, indicating stronger predicted binding affinities and suggesting particularly favorable biochemical interactions within the neuraminidase active site. These findings highlight the considerable potential of C1 and C2 as lead candidates for further therapeutic development. Compound C3, while displaying a docking score lower than Oseltamivir, did not outperform Zanamivir but still demonstrated relevant binding characteristics that justify its inclusion among the most promising hits. All three identified compounds fulfilled the four essential pharmacophoric features established by the e-pharmacophore model, as illustrated in
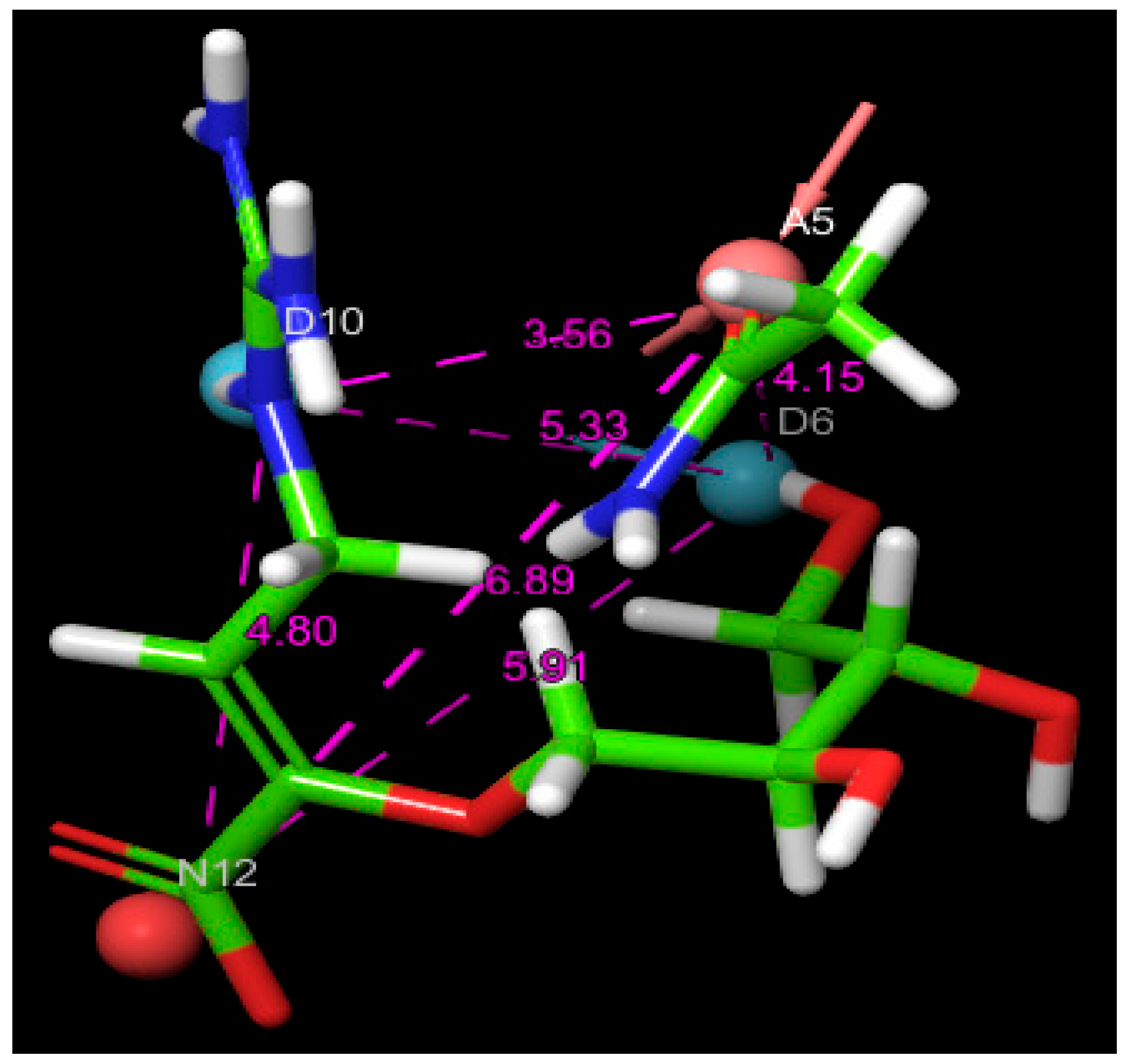
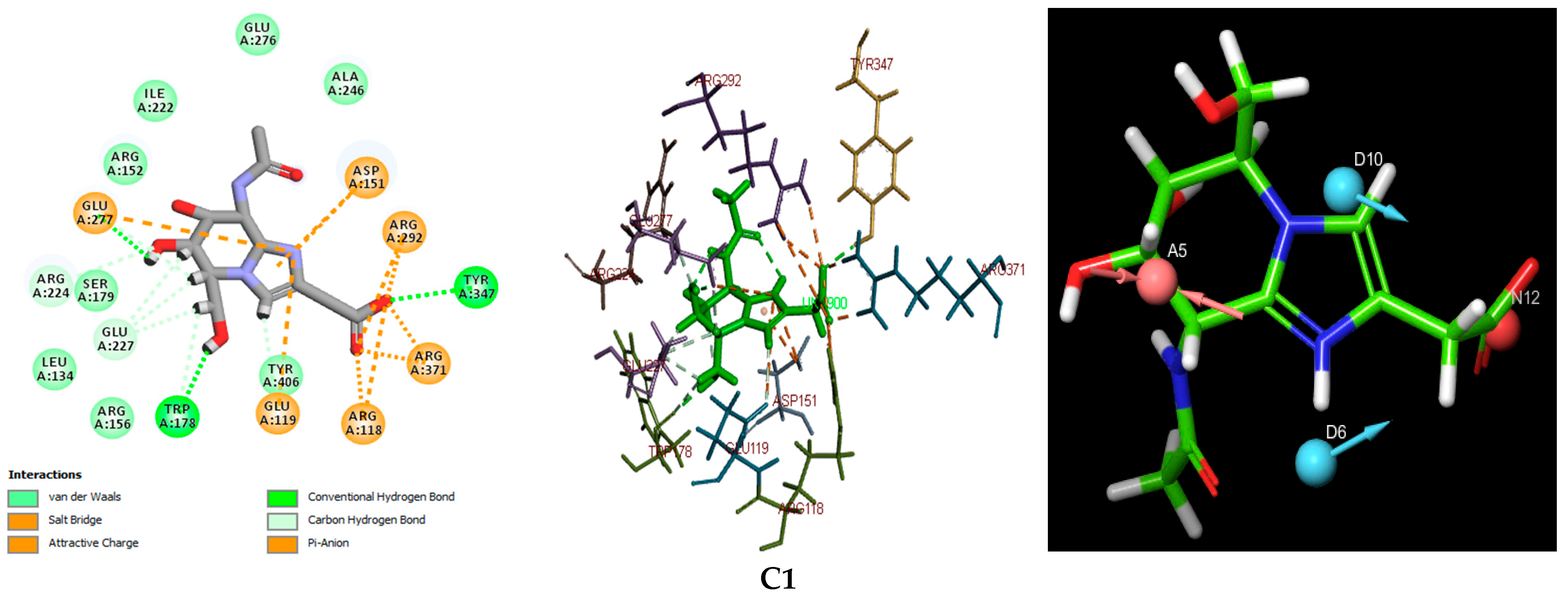
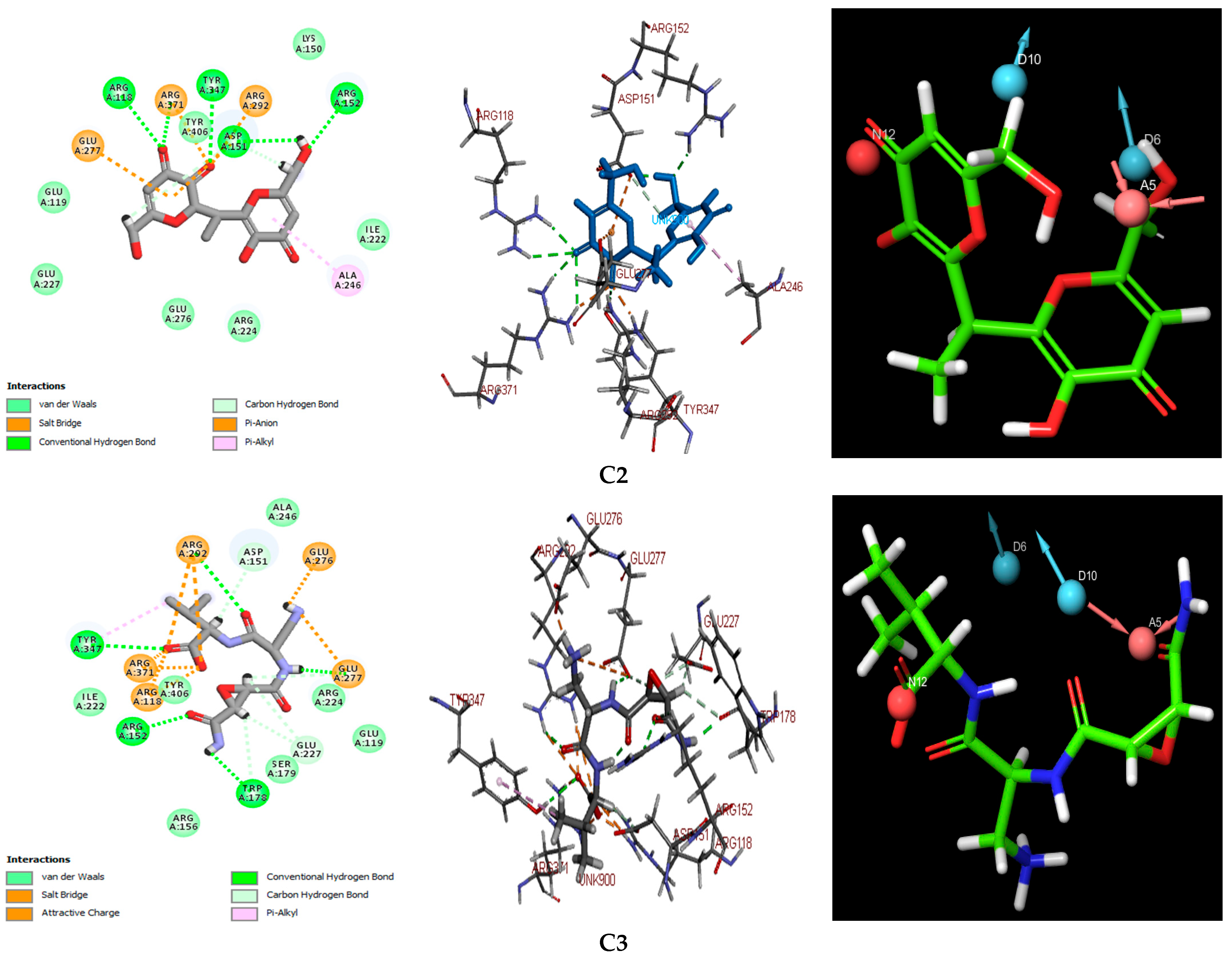
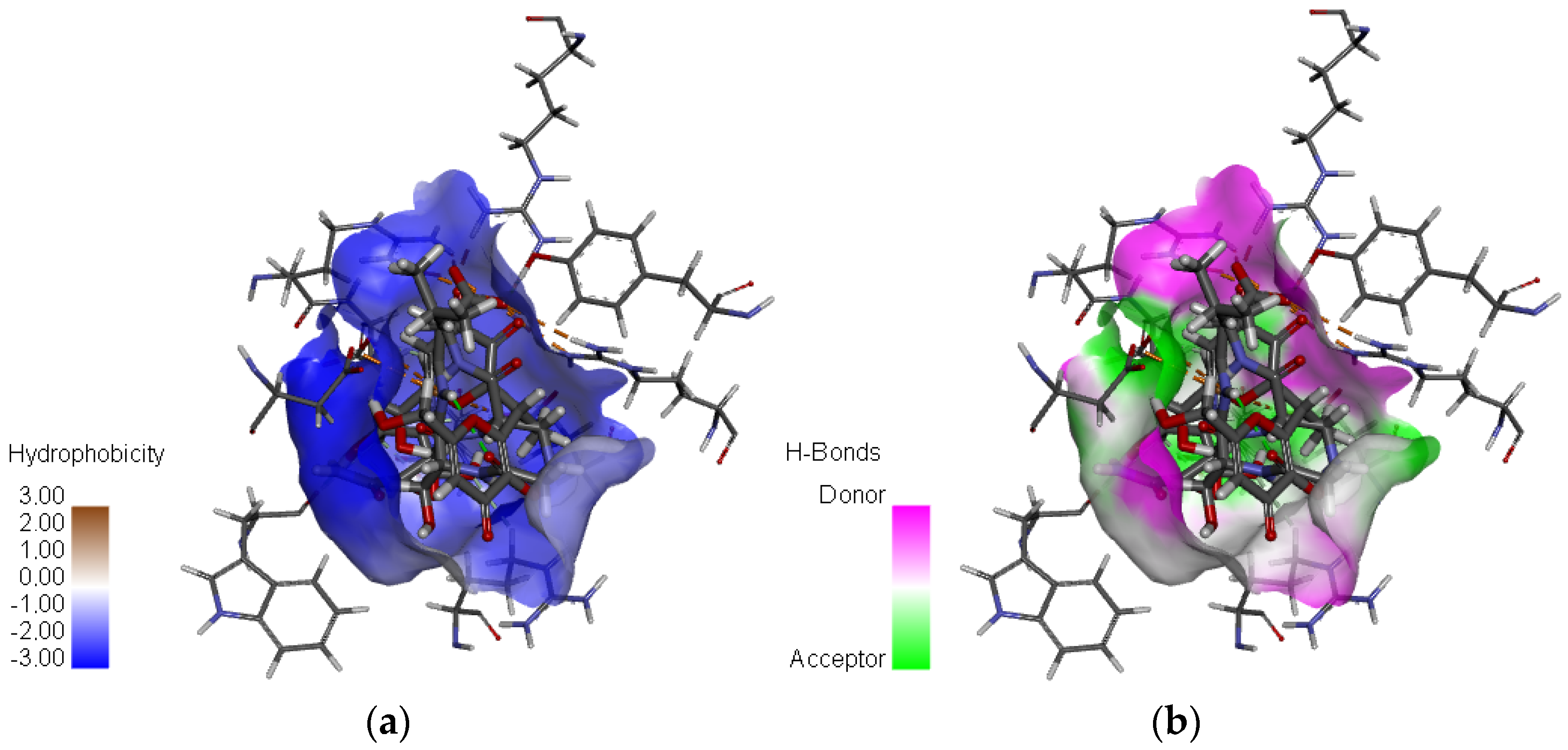
Figure 3. This figure also presents a detailed visualization of the ligand–receptor interactions, enabling a comprehensive interpretation of the docking results. For reference, the binding orientation of Zanamivir within the NA active site is depicted in
Figure 4, offering structural context for comparison with the novel candidates.
The docking results for the three natural compounds, as well as the two antiviral reference inhibitors, Zanamivir and Oseltamivir, are summarized in
Table 3. A comparative analysis of the interaction profiles reveals a striking similarity in the amino acid residues involved in binding between the studied compounds and the reference inhibitors, thus validating the consistency and reliability of our screening approach. A detailed examination of both 2D and 3D interaction maps (presented in
Table 3 and
Figure 3) identified two primary modes of interaction between the compounds and the active site of the H5N1 NA protein (PDB ID: 2HTQ). These modes include hydrogen bond interactions and electrostatic interactions. Both of these interaction types were also observed with the reference inhibitors Zanamivir and Oseltamivir, confirming their critical role in facilitating the binding of the ligands to the protein active site, as shown in
Table 3.
According to the data summarized in
Table 3, the selected compounds demonstrate a significantly higher number of hydrogen bond (H-bond) interactions, with bond distances of less than 3.1 Å, similar to the reference inhibitors. Among the amino acid residues involved in H-bond formation, ARG118, ARG292, ARG371, and TYR347 are identified as key residues due to their direct participation in hydrogen bonding across all ligand–protein complexes analyzed. These interactions are pivotal in the stabilization and specificity of ligand–protein binding, as shown in previous studies [
57,
58,
59,
60,
61].
The results indicate that C1 and C3 form particularly robust biochemical interactions with the target NA protein, marked by a higher number and stability of hydrogen bonds when compared to C2. Additionally, both C1 and C3 exhibit a high number of electrostatic interactions, which are comparable to those observed with Zanamivir. These electrostatic interactions further strengthen the ligand–protein complexes, contributing to their overall binding affinity and specificity [
62,
63]. Among the three compounds, C3 stands out, with the highest XP docking score of −9.433 kcal/mol, and it also exhibits the highest number of hydrogen bonds (18 in total) and electrostatic interactions (10), suggesting the most favorable overall interaction profile with the target protein. The C2 and C3 compounds each form a single hydrophobic interaction, involving the residues ALA246 and TYR347, respectively. While these interactions are limited in number, they are likely to contribute significantly to the binding energy, especially in localized hydrophobic pockets, as illustrated in
Figure 5. These results highlight the complementary role of hydrophobic interactions in stabilizing ligand–protein complexes, particularly when they coexist with more dominant polar and electrostatic interactions [
27,
34]. The key residues identified in our study—Arg118, Arg152, Glu119, Asp151, Trp178, Ile222, Glu227, Arg292, and Arg371—are consistent with those reported in previous studies on neuraminidase inhibitors [
64,
65]. This agreement supports the validity of our docking and pharmacophore models in capturing the essential interactions within the active site of the NA protein (PDB ID: 2HTQ).
3.4. Prediction of ADMET Profiles
The top leads identified through XP molecular docking, using Schrödinger software, were subjected to comprehensive evaluation in ADMET studies. The selection of promising compounds is not solely based on their binding affinity or the nature of their molecular interactions; it also includes crucial pharmacokinetic and therapeutic potential parameters. These factors are vital for assessing the compounds’ efficacy and safety as potential therapeutic candidates [
66].
Table S2 presents the physicochemical and medicinal properties of the compounds under investigation. The results show that the membrane permeability of these molecules is generally limited. Among the compounds, C3 exhibits the highest cell penetration potential, with a TPSA (Topological Polar Surface Area) greater than 150 Å, similar to Zanamivir. In contrast, C1 and C2 have TPSA values ranging from 140 Å to 145 Å, indicating slightly lower cell penetration ability. Notably, all tested compounds, including Zanamivir, have logS values of water solubility within the acceptable range (−4 to 0.5), suggesting adequate aqueous solubility for potential therapeutic use. The logP values for all compounds, including Zanamivir, are negative and fall within the acceptable range (<5), indicating a hydrophilic nature and a low ability to cross the lipid bilayer of cell membranes [
67,
68]. These findings align well with the TPSA values, further confirming the compounds’ limited membrane permeability.
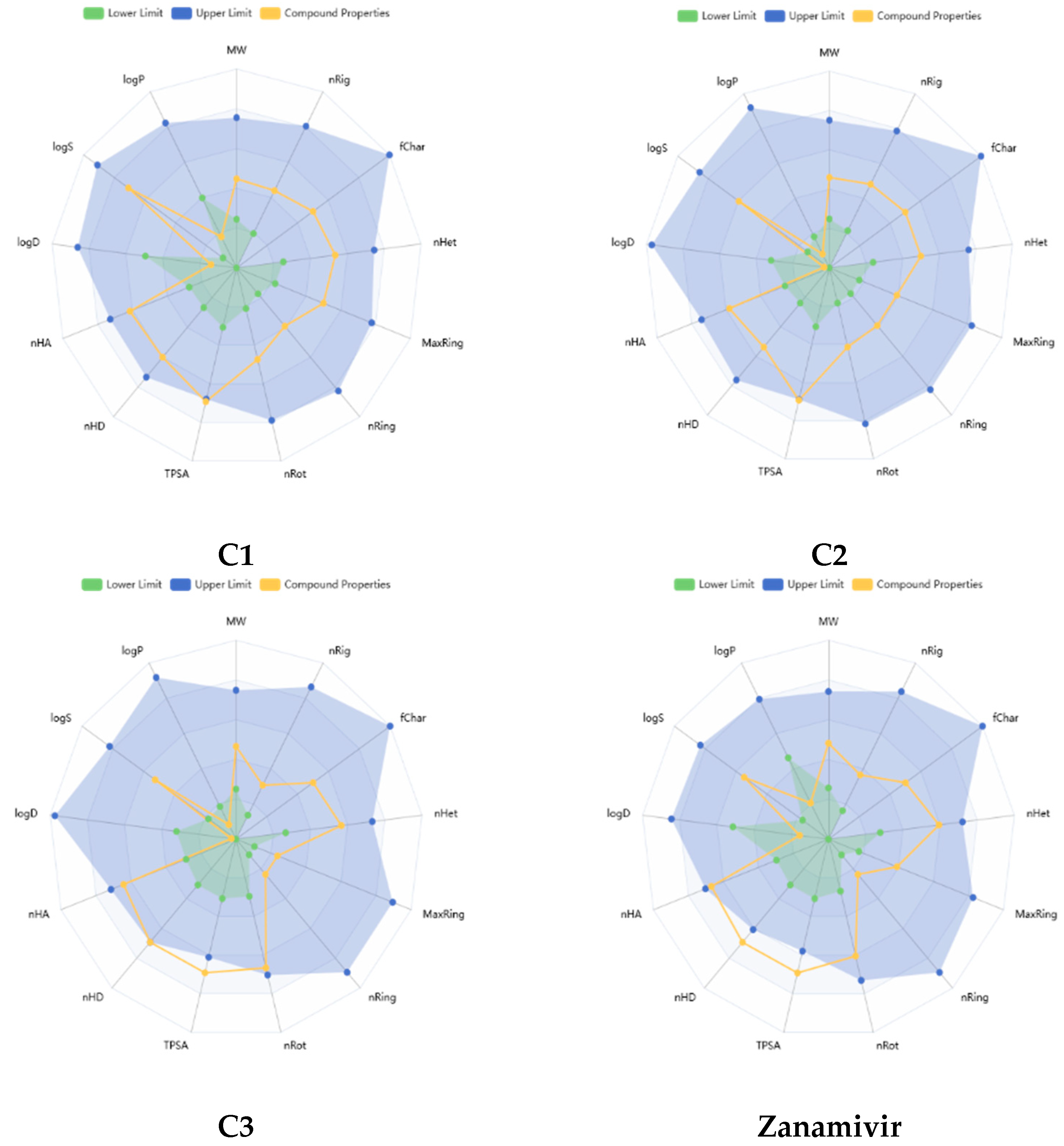
Figure 6 presents a radar chart illustrating the physicochemical properties of the top three compounds (C1, C2, and C3) alongside Zanamivir as a comparison, generated using the ADMET Lab 3.0 server. As shown, the properties of C1 and C2 generally fall within the upper limits (highlighted in blue), indicating favorable profiles. C3, while also performing well, slightly exceeds the recommended range for polarity. In contrast, Zanamivir shows a significant deviation, with TPSA > 140 Å and nHD > 5, indicating that its polarity and hydrogen bond donor count are outside the optimal range for drug candidates.
The results indicate that all of the compounds studied are characterized by easy synthesis as their probability values are all lower than six. Additionally, these compounds fulfill the criteria established by several established drug-likeness rules. Specifically, they meet the Lipinski rule of five [
69] (MW < 500 D, nHA < 10, nHD < 5, logP < 5), the Pfizer rule [
70] (logP < 3, TPSA > 75 Å
2), the GSK rule [
71] (MW < 400 D, logP < 4), and the Golden Triangle rule [
72], all of which indicate optimal bioavailability and strong similarity to known drug molecules. Moreover, no warnings were flagged in the Bristol Myers Squibb (BMS) or Pan Assay Interference (PAINS) rules, suggesting that these compounds have a low risk of generating false positives or misleading results.
Table 4 presents the predictions for the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the three lead compounds and Zanamivir, as analyzed using the ADMETLab 3 software.
Absorption: Concerning the permeation of compounds through human colon adenocarcinoma cell lines (Caco-2), all studied compounds exhibit values lower than −5.15 log cm/s, indicating a low ability to cross the intestinal barrier, which suggests limited absorption. Compound C2 stands out as an exception, showing higher permeability, potentially enhancing its intestinal absorption. The human intestinal absorption (HIA) of compounds C1 and C2 is limited, with values of 15.2% and 5.0%, respectively. In contrast, compound C3 demonstrates a moderate absorption of 62.2%. Zanamivir shows a high absorption rate of 95.8%, indicating excellent bioavailability and optimal intestinal absorption.
Distribution: The distribution of the compounds was evaluated using key parameters, including blood–brain barrier (BBB) permeability, plasma protein binding (PPB), and the volume of distribution (VD). All compounds, including Zanamivir, exhibit probability values close to zero for BBB permeability, suggesting negligible CNS penetration. Since influenza primarily affects respiratory tissues, this limited penetration into the CNS does not interfere with the drug’s action in the most affected areas, such as the lungs [
73]. Regarding PPB, the calculated values for all compounds are below 90%, suggesting a relatively high-unbound fraction in the bloodstream, which may support their availability and therapeutic efficacy. For VD, the predicted values are low (<−0.15), indicating preferential distribution of the compounds in the plasma compartment rather than in tissues.
Metabolism and Excretion: The inhibition of cytochrome P450 enzymes is a key factor in drug metabolism.
Table 4 reveals that none of the tested compounds significantly inhibited the major cytochrome P450 enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, amd CYP3A4), with values ranging from 0 to 0.005, suggesting minimal interaction with the relevant metabolic pathways. In terms of clearance, compounds C1, C3, and Zanamivir show low probabilities (less than 5), indicating reduced clearance, while C2 exhibits a moderate rate of renal clearance (9.627). It is worth noting that low clearance is often desirable in drug discovery as it helps reduce the required dosage while increasing drug exposure [
74]. All compounds display relatively short half-lives (T1/2), ranging from 1.378 to 2.395 h, indicating rapid elimination from the body. Their limited presence in the biological system underscores their rapid excretion.
Toxicity: The ADMETLab 3 results (
Table 4) indicate safe profiles for cardiotoxicity (hERG channel inhibition) and respiratory toxicity. Regarding mutagenicity, the lowest probability values were observed for Zanamivir and C1, with values of 0.209 and 0.394, respectively, indicating a low mutagenic risk for these compounds. In contrast, C2 and C3 display moderate probability values of 0.705 and 0.782, respectively.
Table 5 presents the toxicity predictions from the Protox-II assessment, which evaluates hepatotoxicity, carcinogenicity, mutagenicity, and cytotoxicity. Similar to Zanamivir, the top three leads show favorable safety profiles, with no significant risks for hepatotoxicity, carcinogenicity, mutagenicity, or cytotoxicity. These findings suggest that these compounds pose limited toxicological risks, enhancing their potential for therapeutic or industrial applications.
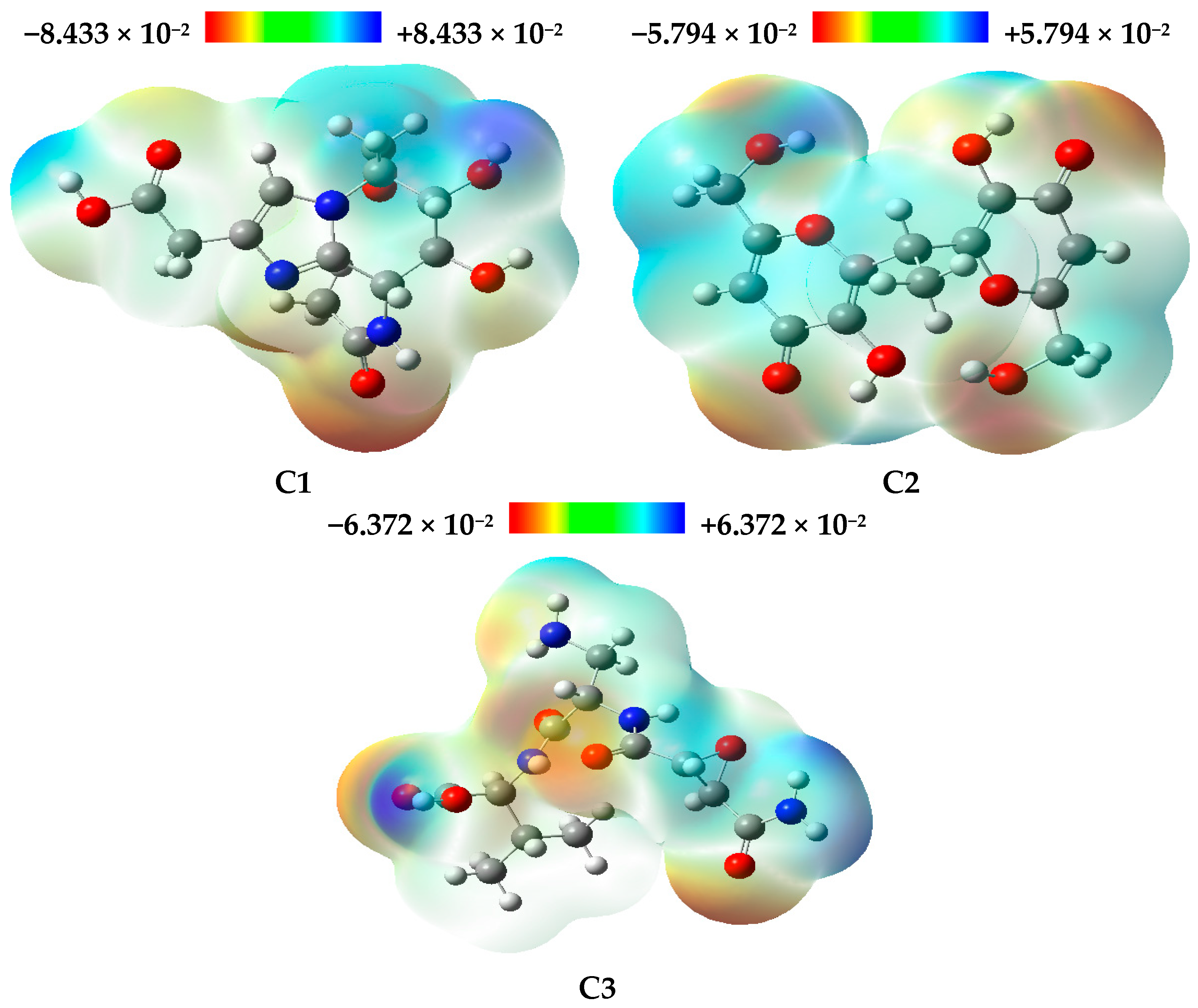
3.5. Frontier Molecular Orbitals Analysis and Global Reactivity Descriptors
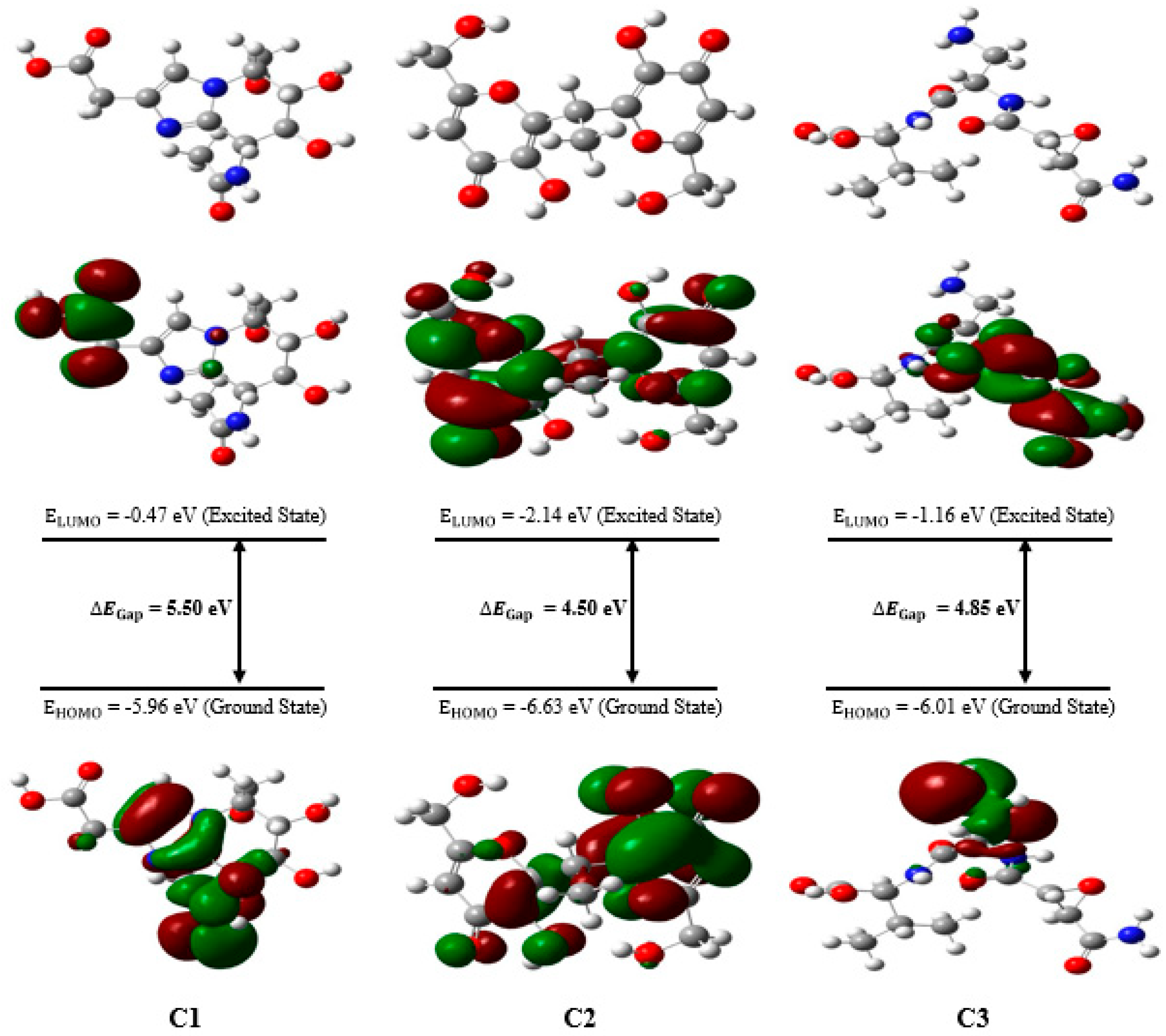
The global reactivity descriptors for the three natural compounds studied, namely C1, C2, and C3, were obtained through DFT calculations. The values for each compound, as summarized in
Table 6, provide insights into their electronic properties and reactivity. These descriptors are important for understanding the chemical behavior and stability of the compounds, which are crucial for evaluating their potential as therapeutic agents.
The results reveal that compound C1 has the highest value of E
HOMO, suggesting increased reactivity toward electrophiles. On the other hand, compound C2 exhibits the lowest E
LUMO value, indicating a greater tendency to interact with nucleophiles.
Figure 7 displays the optimized structures of the investigated compounds, highlighting the HOMO and LUMO molecular orbitals, as well as their corresponding energy gaps. In this figure, the red and green colors represent the positive and negative phases of the molecular orbitals, respectively.
The energy gap (
) is a crucial parameter for predicting the reactivity and chemical stability of a compound. The high value of
observed for compound C1 indicates greater chemical stability compared to the other compounds studied. Indeed, a larger
between the boundary molecular orbitals, such as HOMO and LUMO, is typically associated with better thermodynamic and chemical stability as it makes electronic transitions more difficult. In contrast, compounds C2 and C3, which exhibit a smaller energy gap, are expected to have increased reactivity and lower stability. A smaller
facilitates electronic transitions and interactions with other chemical species, thereby enhancing their chemical reactivity.
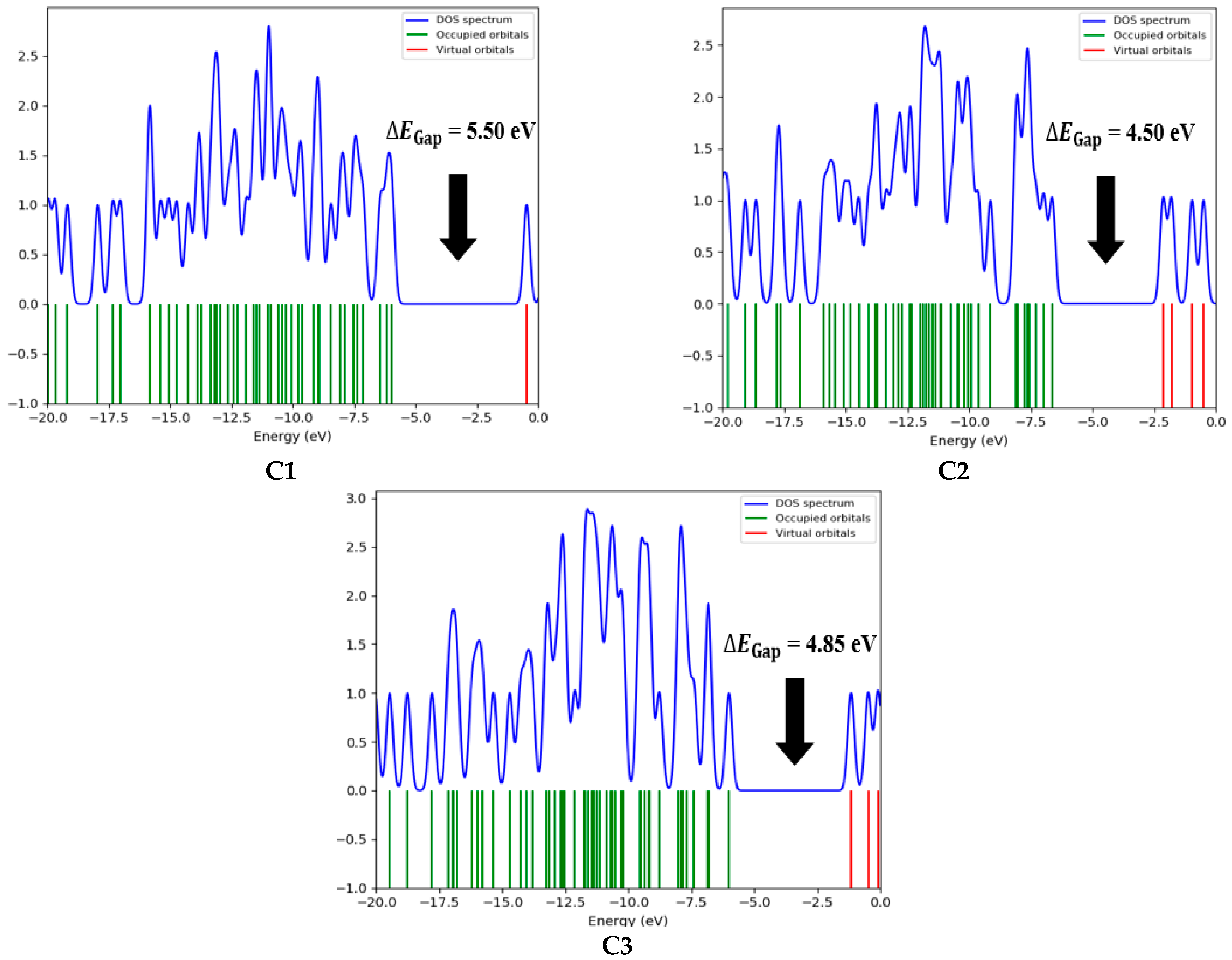
Figure 8 presents the DOS spectrum generated from the Gauss Sum program for the three natural compounds, confirming the calculated
values.
The concepts of chemical hardness and softness are fundamental in assessing both the stability and reactivity of molecules. Compound C2, with the lowest chemical hardness (η = 2.25 eV) and the highest chemical softness (S = 0.22 eV), is the most reactive of the compounds studied. Compound C1 is considered the most stable of the three as it has the highest value of chemical hardness (η = 2.75 eV), which also contributes to its greater stability. Chemical potential (μ) corresponds to the tendency of a system to donate electrons, and a negative chemical potential reflects the stability of the complex, meaning it does not spontaneously decompose into its constituents [
75].
Table 6 shows that compound C2 has the lowest chemical potential (μ) and the highest electronegativity (χ), suggesting that it functions as a strong electron acceptor. In contrast, compound C1 has the highest chemical potential, implying that it could interact more readily with the environment to exchange electrons.
The electrophilicity index (ω) is a key quantum parameter used to quantify the chemical reactivity of molecules. It represents the ability of a chemical species to accept electrons, evaluating its potential for interaction with biological receptors, particularly in drug design. A low value of ω characterizes a good nucleophile, making it more reactive, while a high value indicates a powerful electrophile [
75,
76]. Based on the results in
Table 6, compound C2 exhibits the highest reactivity, with a ω value greater than 4 eV, followed by compound C3, with an electrophilicity index of 2.65 eV. Compound C1, characterized by a high nucleophilicity index (N) and the lowest value of ω, stands out as the most nucleophilic compound among the studied compounds. Notably, the HOMO energy of the reference system (TCE) was determined to be −9.16 eV using the DFT/B3LYP method with the 6-31G basis set [
43].
3.7. MD Simulations
In this study, molecular dynamics (MD) simulations were performed to investigate the structural behavior and interaction profiles of four ligand–protein complexes involving the neuraminidase (NA) enzyme of avian influenza H5N1 (PDB ID: 2HTQ). The tested ligands included three candidate compounds (C1, C2, and C3) and the reference antiviral drug Zanamivir. The resulting complexes—C1-NA, C2-NA, C3-NA, and Zanamivir-NA—were analyzed over a 100 ns simulation period to assess their conformational stability, residue flexibility, and interaction continuity within the active site of the NA protein. The following sections present a comparative analysis of the RMSD, RMSF, and residue–ligand contact maps to highlight the dynamic performance of each complex.
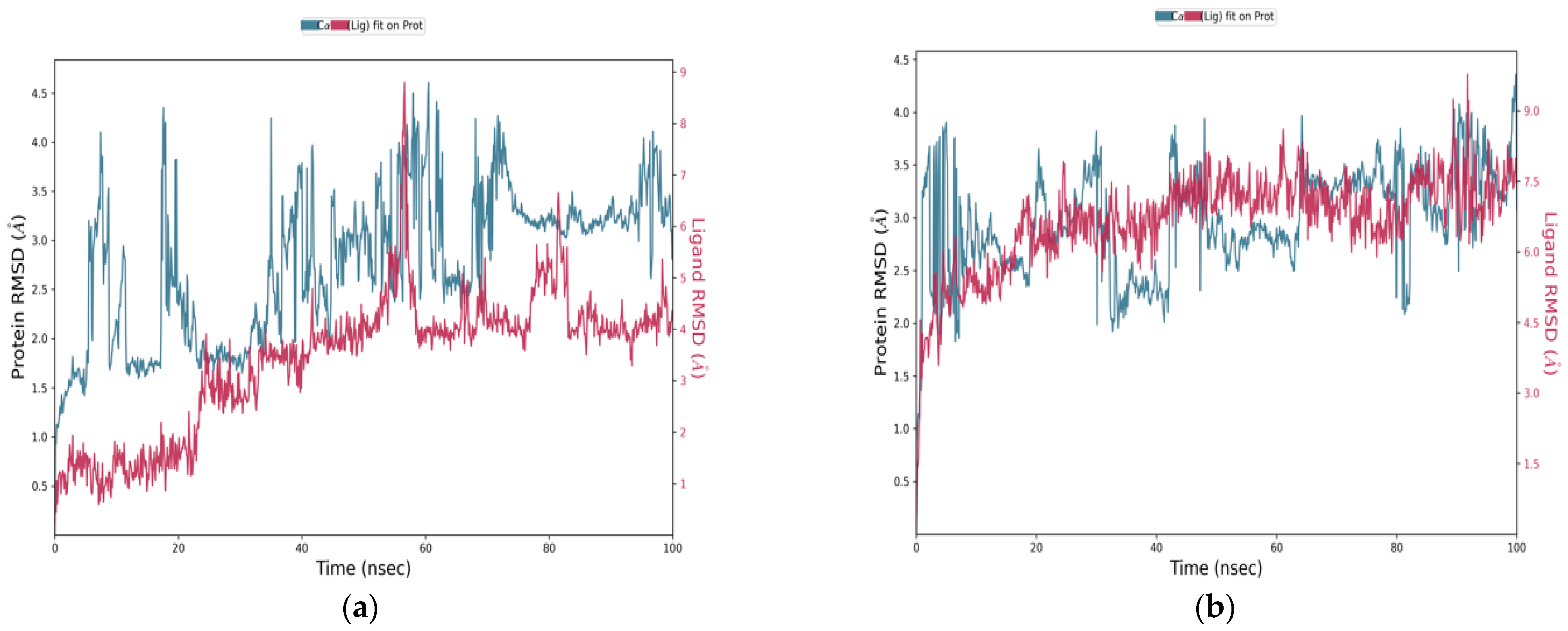
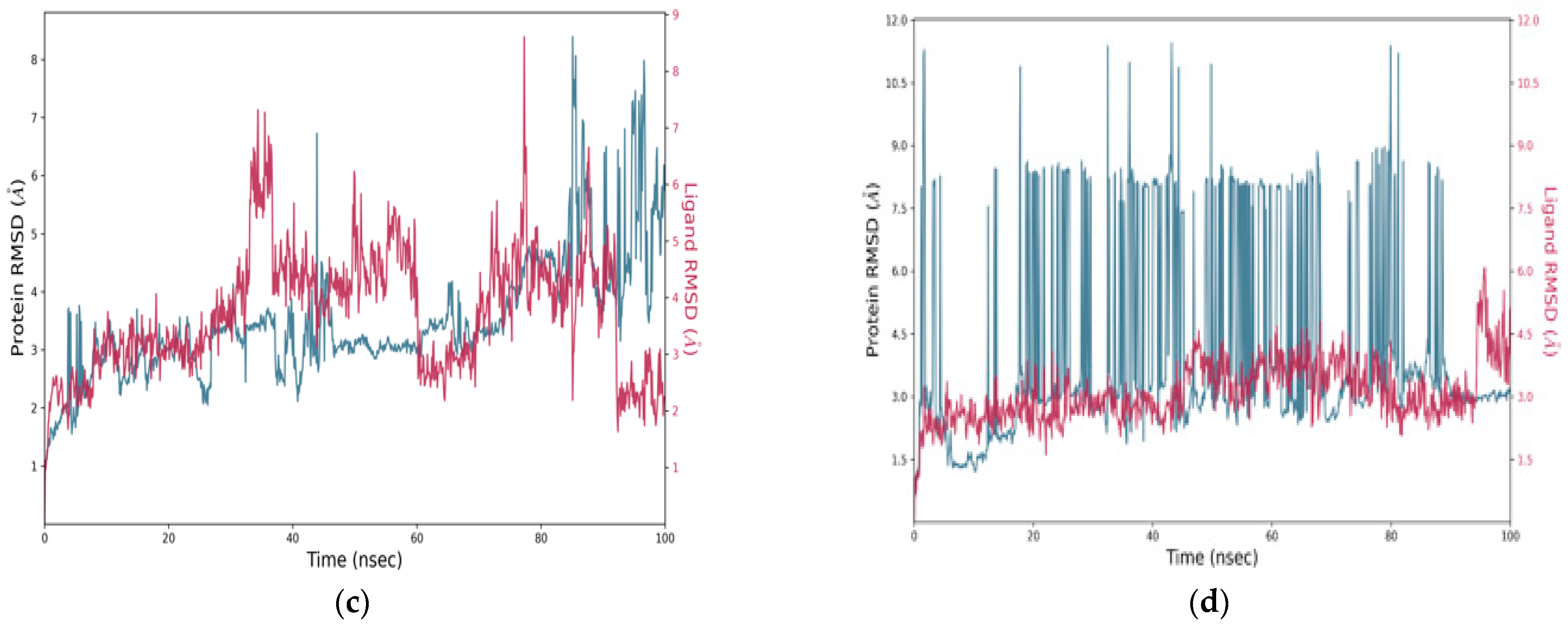
The RMSD plots over 100 nanoseconds (
Figure 10) reveal significant differences in the behavior of the candidate compounds compared to the reference ligand, Zanamivir. These differences reflect variations in structural stability and anchoring within the active site. The C1-NA complex (
Figure 10a) showed an initial fluctuation in the protein’s RMSD, reaching about 2.5 Å, followed by stabilization around 3.0–3.5 Å, indicating structural reorganization followed by a stable phase. The ligand’s RMSD remained generally below 5 Å, suggesting relative stability in the binding site. For the C2-NA complex (
Figure 10b), the protein’s RMSD remained in the range of 2.5–3.5 Å without abrupt variations, reflecting higher global stability. The ligand’s RMSD was relatively stable around 6.5–7.5 Å, indicating consistent and maintained positioning throughout the simulation. The C3-NA complex (
Figure 10c) showed RMSD fluctuations between 2.5 and 4.0 Å, with a tendency toward stabilization in the second half of the simulation. The ligand maintained an RMSD between 4.5 and 6.5 Å, with a noticeable improvement in the latter phase, indicating progressive adaptation to the active site. In contrast, the Zanamivir-NA complex displayed unstable behavior, with the protein RMSD ranging from 2.0 to 11.0 Å, accompanied by frequent and marked fluctuations (
Figure 10d). The ligand RMSD occasionally exceeded 8.0 Å, indicating considerable structural instability and potential partial disengagement from the binding site. These results suggest that C1 and C2, in particular, exhibit superior dynamic stability compared to the reference molecule, reinforcing their potential as promising therapeutic candidates.
To complement the RMSD analysis and gain deeper insights into the flexibility and interaction patterns of the ligand–protein complexes, further investigations were carried out through root mean square fluctuation (RMSF) profiles and interaction mapping.
C1 formed a diverse array of interactions distributed across several active site residues (
Figure 11a). These included strong water bridges, notably with GLU277 (interaction fraction > 1.6) and with ARG152, ARG118, GLU119, and ASP151 (values between 0.5 and 1.1). Clear ionic bonds were also observed, particularly with GLU277, GLU119, and ARG371, indicating solid electrostatic interactions between oppositely charged groups. Furthermore, the ligand formed notable hydrogen bonds with ASP151, ARG152, LYS151, and ARG371. Hydrophobic interactions, though present, were limited and involved primarily ARG156, LEU134, and TYR406 at relatively low frequency. This interaction profile reflects an effective three-dimensional anchoring of the ligand in the active site, supported by a combination of water bridges and ionic interactions. This aligns well with the low RMSD of the ligand (<5 Å) and the protein complex stability (3–3.5 Å), corroborated by high docking performance.
C2 established specific interactions with several key residues in the active site (
Figure 11b). Significant ionic bonds were formed, particularly with GLU227, GLU119, and ARG151, although frequencies varied. Clear hydrogen bonds were observed with TYR347 and GLU277, indicating directional anchoring in the catalytic site. Additionally, water bridges were detected between the ligand and ASP198 and ARG152, with an interaction frequency around 0.6, further strengthening complex stability. Hydrophobic interactions involving ILE222, TRP178, and ARG152 suggested ligand insertion into a nonpolar region of the active site. Despite a relatively high ligand RMSD (6.5–7.5 Å), the combination of electrostatic and hydrophobic interactions allowed for prolonged and relatively stable ligand retention. This stability may be attributed to a balanced interaction profile between polarity and flexibility, explaining the ligand’s persistence in the active site throughout the simulation.
C3 showed a particularly rich and diverse interaction profile (
Figure 11c). It formed strong hydrogen bonds, notably with ARG292 (interaction frequency > 1.4), and important water bridges with essential residues such as GLU277, ASP151, ARG152, GLU119, and ARG371, reinforcing ligand integration into the active site. Ionic bonds were well represented, involving GLU227, ASP151, GLU119, and ARG371, supporting effective electrostatic stabilization. Additionally, notable hydrophobic interactions with TYR347 and TYR406 suggested favorable positioning in nonpolar binding pockets. This diversity of interactions highlights C3′s ability to simultaneously exploit different molecular forces to ensure robust binding within the active site. This is consistent with a moderate ligand RMSD (4.5–6.5 Å) and a notable improvement in structural stability in the second half of the simulation, indicating good adaptive flexibility of the ligand–protein complex.
The reference compound Zanamivir initially exhibited strong interactions with several key residues in the active site (
Figure 11d). It formed marked hydrogen bonds with GLU277 (interaction frequency ~1.3), as well as with GLU119, ASP151, and ARG152 (0.5–1.0 range). It also showed notable reliance on ionic interactions, mainly with ARG152 and GLU277, with minor contributions from GLU119 and ASP151. Water bridges were also observed, particularly with ARG152, GLU277, ARG292, and GLU119, reflecting water involvement in complex stabilization. However, there was a near absence of hydrophobic interactions, limiting the ligand’s overall stability in a dynamic environment. This imbalance in interaction types may explain the marked fluctuations observed in the molecular simulation, where the protein RMSD exceeded 10 Å and the ligand RMSD surpassed 8 Å. Despite strong initial affinity, Zanamivir exhibited significant long-term structural instability, likely due to a lack of complementarity with the nonpolar regions of the active site.
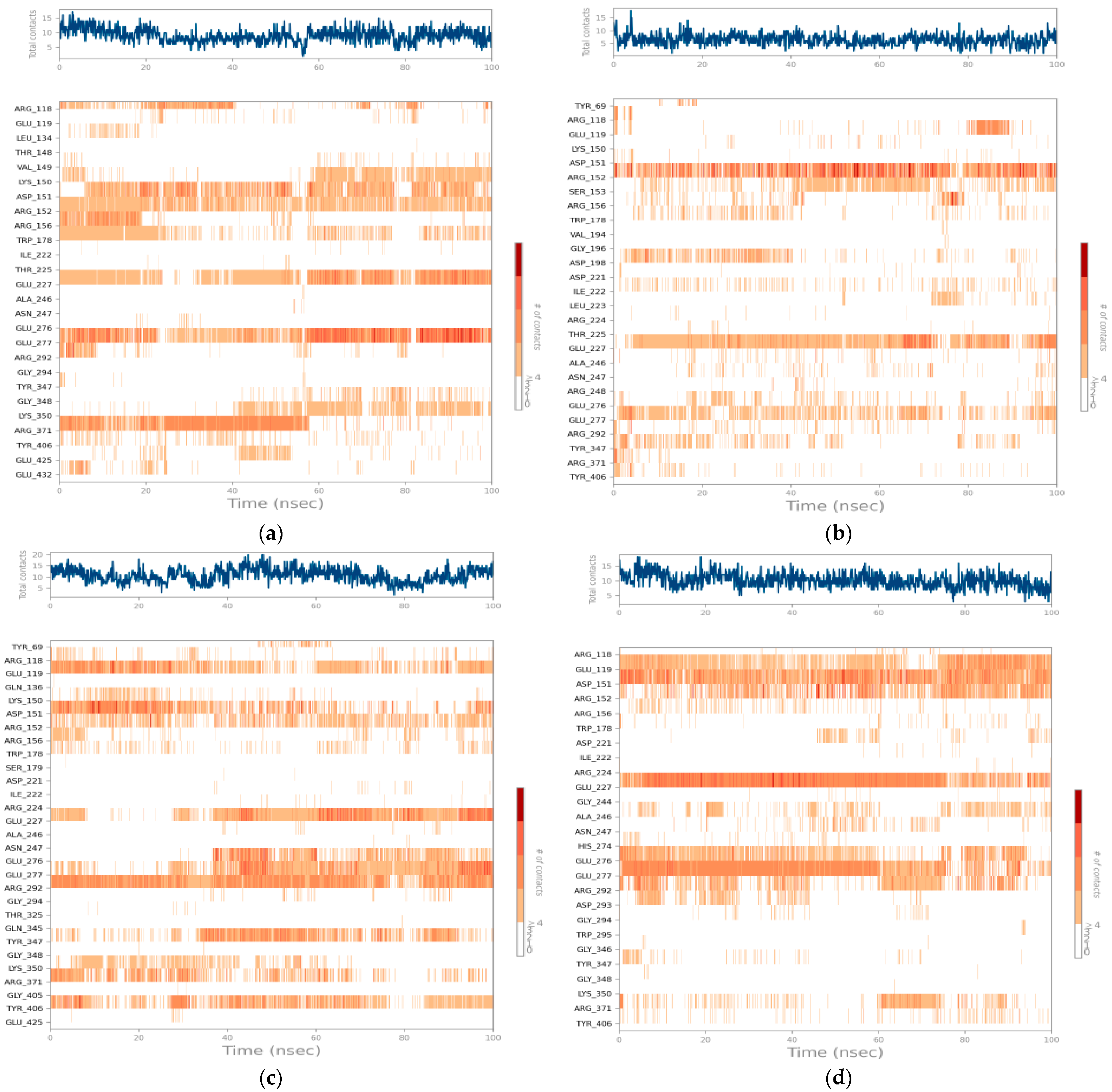
An analysis of the contact maps during the 100-nanosecond molecular simulation (
Figure 12) revealed notable differences in ligand behavior in terms of stability and the persistence of interactions with active site residues.
Compound C1 (
Figure 12a) exhibits a high and relatively constant interaction density, fluctuating between 10 and 15 contacts throughout the trajectory. This profile reflects remarkable stability of the complex, with residues such as ARG224 and GLU277 maintaining long-lasting contacts throughout the simulation. Sustained interactions are also observed with ARG152, ASP151, and ARG292, reinforcing the idea of deep, multiregional ligand anchoring. The consistency in these interactions supports the low RMSD values and confirms the stability of the complex over time.
Compound C2 (
Figure 12b) displays more fluctuating dynamics, with a total number of contacts ranging from 5 to 12. The most frequent interactions involve ARG151, SER153, GLU277, and ALA246, but these interactions are more intermittent and less persistent over time. This partial instability could explain the increased mobility of the ligand in the active site, as revealed by the higher RMSD values observed previously. The fluctuation in the number of contacts suggests that C2 is not as firmly anchored in the active site as C1, leading to greater mobility and reduced stability.
Compound C3 (
Figure 12c), on the other hand, demonstrates intermediate but very promising behavior. The number of contacts fluctuates, sometimes reaching nearly 18, with a strong intensity in the second half of the simulation. C3 establishes consistent interactions with key residues, including GLU277, ARG292, GLU119, and ARG371. This prolonged bond retention, illustrated by strong horizontal bands on the map, demonstrates excellent structural complementarity and a good dynamic fit within the enzymatic site. The high number of contacts, particularly in the latter half of the simulation, highlights C3’s ability to stabilize itself through consistent and diverse interactions.
Zanamivir (
Figure 12d), in contrast, initially displayed a numerically similar interaction profile to C1, but a detailed analysis of the interaction map revealed a lack of continuity. Frequent breaks in interactions, even at critical residues such as ARG152, GLU277, and ARG224, suggest gradual destabilization over time. This lack of interaction stability explains the large variations in dynamic parameters such as the RMSD and RMSF despite its initial binding energy being comparable to that of the candidate compounds.
In contrast to Zanamivir, complexes C1-NA, C2-NA, and C3-NA exhibited better dynamic stability. The C1-NA complex is characterized by rich, balanced interactions and a low ligand RMSD deviation, confirming its structural stability throughout the simulation. The C2-NA complex maintained its stability despite greater mobility, thanks to a combination of ionic and hydrophobic interactions. The C3-NA complex, incorporating a variety of ligand types, adapted effectively within the active site, showcasing excellent flexibility and stability in the latter part of the simulation.
Overall, the candidate compounds C1, C2, and C3 displayed better dynamic stability compared to Zanamivir, which suffered from a lack of interaction diversity, particularly the absence of hydrophobic bonds. This imbalance likely contributed to Zanamivir’s lower stability over time despite its initial strong binding.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}