1. Introduction
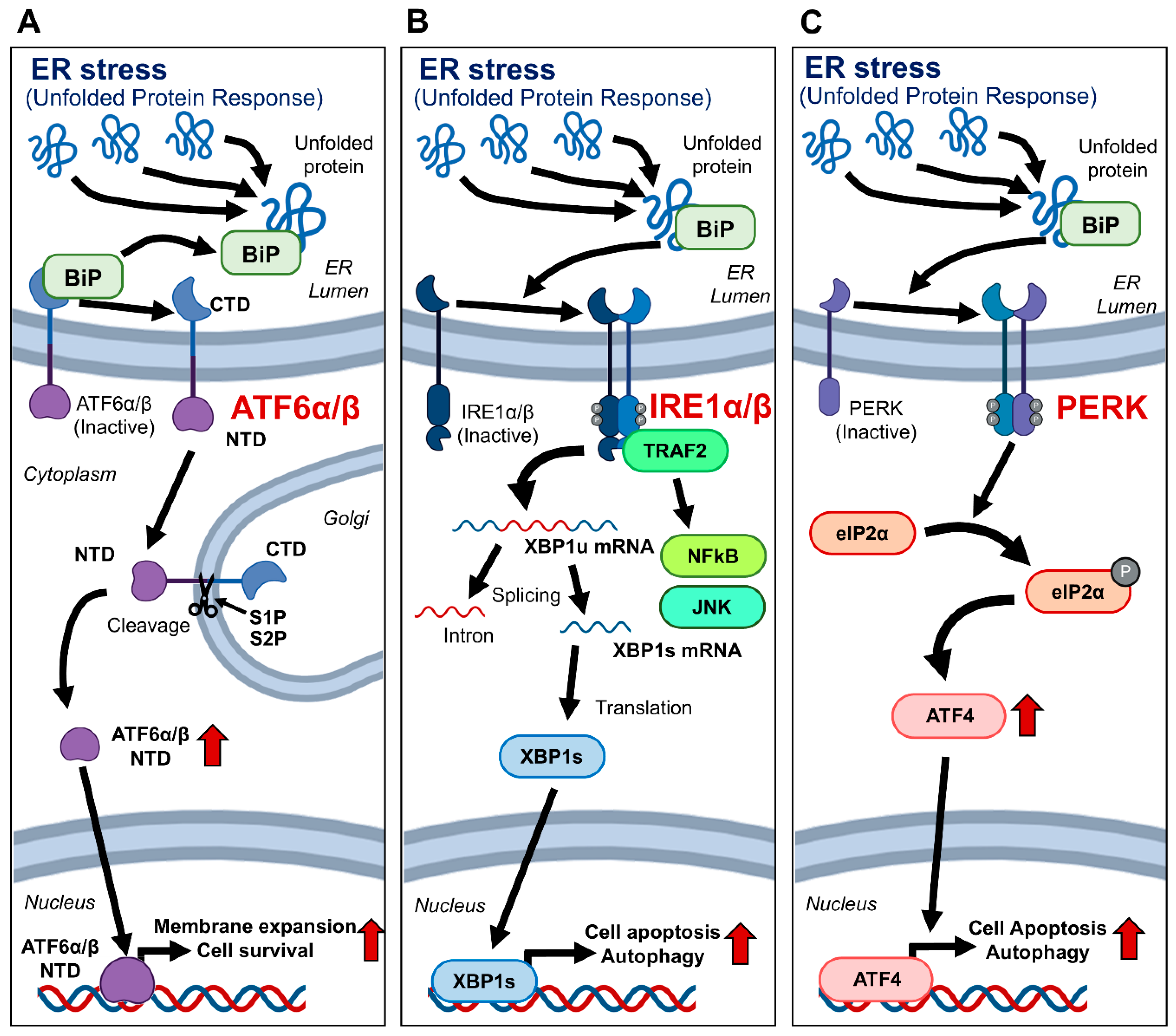
The unfolded protein response (UPR) is an essential cellular mechanism activated in response to the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) [
1,
2,
3]. This process is critical for maintaining cellular homeostasis and proper protein folding. The UPR is primarily regulated by three ER stress sensors: ATF6, IRE1, and PERK [
1,
3]. Among these, ATF6, which includes ATF6A and ATF6B isoforms, plays a central role by translocating to the Golgi upon ER stress, where its active form is generated to initiate transcriptional responses [
4,
5]. Among the UPR regulators, ATF6B has emerged as a potential target for modulating membrane protein production due to its distinct role in ER stress adaptation.
In the Golgi, ATF6α/β undergoes cleavage by Site-1 and Site-2 proteases, releasing its N-terminal cytoplasmic domain [
6,
7]. This domain acts as a transcription factor, upregulating genes that enhance the protein-folding capacity of the ER and degrade misfolded proteins, thus mitigating ER stress. IRE1α/β normally exists as a monomer and dimerizes upon activation by unfolded proteins [
8,
9]. This dimerization recruits TRAF2, activating NFκB and JNK signaling pathways [
10,
11]. Additionally, IRE1α/β splices XBP1 mRNA to produce XBP1s, which acts as a transcription factor to upregulate ER stress response genes [
12]. PERK also plays a significant role in the UPR by dimerizing upon ER stress activation. This dimerization phosphorylates eIF2α, leading to a reduction in general protein synthesis, which alleviates the burden on the ER [
5,
11]. In addition, it enhances the translation of ATF4, inducing the expression of genes involved in amino acid metabolism, redox reactions, and apoptosis, thereby contributing to cell survival under stress conditions [
13] (
Figure 1).
HEK293T cells are extensively used in the bioindustry for the production of recombinant proteins and viral vectors [
14,
15,
16]. Their human origin makes them particularly suitable for producing biologics intended for human use, ensuring more accurate post-translational modifications and reducing immunogenicity compared to non-human cell lines like CHO cells [
14]. These characteristics make HEK293T cells ideal for producing therapeutic proteins and viral vectors used in gene therapy, vaccine development, and cancer treatments [
15,
16,
17]. However, despite these advantages, HEK293T cells may present limitations such as inconsistent protein expression and challenges in scalability for large-scale production. Among the proteins obtained from HEK293T cells, membrane proteins are of higher industrial value than cytosolic proteins due to their easier separation and purification process [
18,
19]. Given the high costs associated with purification, improving the productivity of membrane proteins derived from HEK293T cells is essential for their broader use in the industry [
14,
18,
20]. Various strategies, including transient transfection, stable cell line development, and site-specific integration using recombinase-mediated cassette exchange, have been employed to enhance protein production in HEK293T cells [
20,
21,
22,
23,
24]. Despite these advancements, challenges such as scalability, reproducibility, and the complexity of achieving stable high-yield production persist.
Since the UPR is an essential process for cell survival, particularly under stress conditions, numerous investigators revealed that this mechanism allows cells to minimize membrane protein production and other non-essential functions, thereby focusing resources on managing stress and promoting cell survival [
1,
11,
25,
26,
27]. Instead of completely blocking the UPR, selective modulation of specific components, such as ATF6B, may help alleviate excessive stress responses and improve membrane protein production. In this study, the C-terminal domain of ATF6B—a regulatory element involved in controlling UPR intensity—was selectively deleted using CRISPR-Cas9 technology. This deletion was found to prevent overactivation of the UPR without inducing cytotoxicity, thereby establishing a cellular environment more favorable for membrane protein biosynthesis.
2. Materials and Methods
2.1. Cell Culture and Transfection
HEK293T cells were obtained from the Korean Cell Line Bank (Seoul, Republic of Korea), which also provided short tandem repeat (STR) profiling data to authenticate the cell line. The STR markers were as follows: D3S1358 (15,17), TH01 (7,9.3), D21S11 (28,30.2), Penta_E (7,15), D5S818 (8,9), D13S317 (12,14), D7S820 (11), D16S39 (9,13), CSF1PO (11,12), Penta_D (9,10), Amelogenin (X), vWA (16,19), D8S1179 (12,14), TPOX (11), FGA (23), D19S433 (18), and D2S1338 (19). The full STR profile is provided in
Supplementary Table S1. Cells were maintained in Dulbecco’s Modified Eagle′s Medium (DMEM; Hyclone, Locan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 50 units/mL penicillin, and 50 μg/mL streptomycin (GibcoBRL, Carlsbad, CA, USA) at 37 °C in humidified air containing 5% CO
2. All plasmid DNA transfections in HEK293T cells were conducted using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA), which was chosen for its high transfection efficiency and cell viability in HEK293T cells, as well as its compatibility with previously established protocols in our laboratory.
2.2. Construction of Plasmid DNA Vectors
The All-in-One vector system (named pL-CRISPR.EFS.tRFP), kindly provided by Benjamin Ebert (Addgene plasmid # 57819;
http://n2t.net/addgene:57819; accessed on 21 June 2022; RRID:Addgene_57819), was utilized as the starting material (
Table 1). To produce a stable cell line, a lentivirus vector of two vector systems was produced. First, to create a single guide RNA (sgRNA) expression vector named pL-sgRNA.EFS.Zeo.tRFP, the Cas9 gene was excised from the pL-CRISPR.EFS.tRFP vector, and a bleomycin resistance gene was inserted. Additionally, a lentivirus vector expressing Cas9 and neomycin resistance genes was synthesized using VectorBuilder (Guangzhou, China). A lentiviral vector facilitating the stable expression of a membrane-bound form of green fluorescent protein (mGFP) was engineered by cloning the GAP43 palmitoylation signal peptide sequence along with GFP into the pLVX-CMV-IRES-Puro vector (manufactured by Takara Bio, Inc., Otsu, Japan). The structural design and component organization of the lentiviral plasmids used in this study are illustrated in
Figure 2A,B, which depict the Cas9/sgRNA expression vector and the sgRNA-only vector, respectively. These schematics are provided to enhance clarity regarding functional elements and selection markers.
This is a list of primers used to create CRISPR-Cas9 constructs for candidate genes. (For cloning, forward and reverse primers were annealed together, and then ligation was performed using T4 ligase).
2.3. Design and Construction of sgRNAs Targeting Candidate Genes
Utilizing the CRISPR RGEN Tool’s Cas-Designer (
http://www.rgenome.net/cas-designer/, accessed on 21 June 2022), sgRNAs targeting ATF6A, ATF6B, IRE1A, IRE1B, and PERK were designed (one set of 3 sgRNAs for each target gene); ATF6A and ATF6B remove the C-terminal transmembrane domain (TMD), IRE1A and IRE1B suppress the expression of genes, and PERK removes the cytoplasmic domain (CPD) and protein kinase domain (PKD) at the C terminus. To create the designed sgRNA expression vector for genetic screening, oligonucleotide pairs encoding each sgRNA were synthesized through Bionics (Seoul, Republic of Korea). These oligonucleotide pairs were inserted into pL-CRISPR.EFS.tRFP to create a construct expressing each sgRNA. To produce stable cells, ATF6B-TMD-1 was inserted into the pL-sgRNA.EFS.Zeo.tRFP vector. The sgRNA targeting the transmembrane domain (TMD) of ATF6B was designed to induce deletion of the C-terminal region. Cells edited using this sgRNA are referred to as “ATF6B-ΔC” throughout this manuscript.
2.4. T7 Endonuclease I Assay (T7E1 Assay)
To evaluate the activity of each sgRNA, HEK293T cells were transfected with All-in-One vectors expressing each sgRNA. After 3 days of transfection, 5 × 106 transfected cells were harvested, and the genomic DNA of each sample was purified using the G-DEX™ IIc Genomic DNA Extraction Kit (iNtRON Biotechnology, Seoul, Republic of Korea). Genomic DNA purification was performed according to the manufacturer’s instructions.
To obtain the amplified target gene PCR product for each sample, target regions were amplified through PCR (EzPCR™ HF 5x PCR Master Mix; Elpis Biotech, Daejeon, Republic of Korea) using the designed primer pair (
Table 2). The first step of PCR conditions was 1 cycle of 5 min at 95 °C, followed by 10 cycles of 30 s at 95 °C, 30 s at 70 °C (annealing temperature was decreased by 1 °C per cycle to 60 °C), and 2 min at 72 °C. Next, the second step of PCR conditions was 30 cycles of 30 sec at 95 °C, 30 sec at 60 °C, and 2 min at 72 °C, finishing with 3 min incubation at 72 °C. After amplification of target genes, the hybrid DNA duplex is formulated by PCR cycling method as follows: 2 min at 95 °C, ramp down (−2 °C/s) at 95 °C to 85 °C, and (−0.1 °C/s) at 85 °C to 25 °C. For the T7E1 reaction, a 20 µL mixture (containing 2 µL of 10× NEB buffer-2, 10 units of T7E1 enzyme, and 10 µL of hybrid DNA duplex) was prepared, and the reaction was carried out at 37 °C for 20 min. T7E1 reactants were separated on a 1.5% agarose gel in TAE buffer containing 1× RedSafe Nucleic Acid Staining Solution (iNtRon Biotechnology) by electrophoresis and visualized under a Blue LED light.
This is a list of PCR primers used to analyze the results of candidate genes. These are T7E1 primers for PCR amplification of the target region of each candidate gene used in the T7E1 assay and primers for NGS for edited sequence analysis of ATF6B-TMD (ATF6B-ΔC), which is the final candidate.
2.5. Generation of Stable HEK293T Cell Lines with Cas9, mGFP, and Targeted sgRNAs
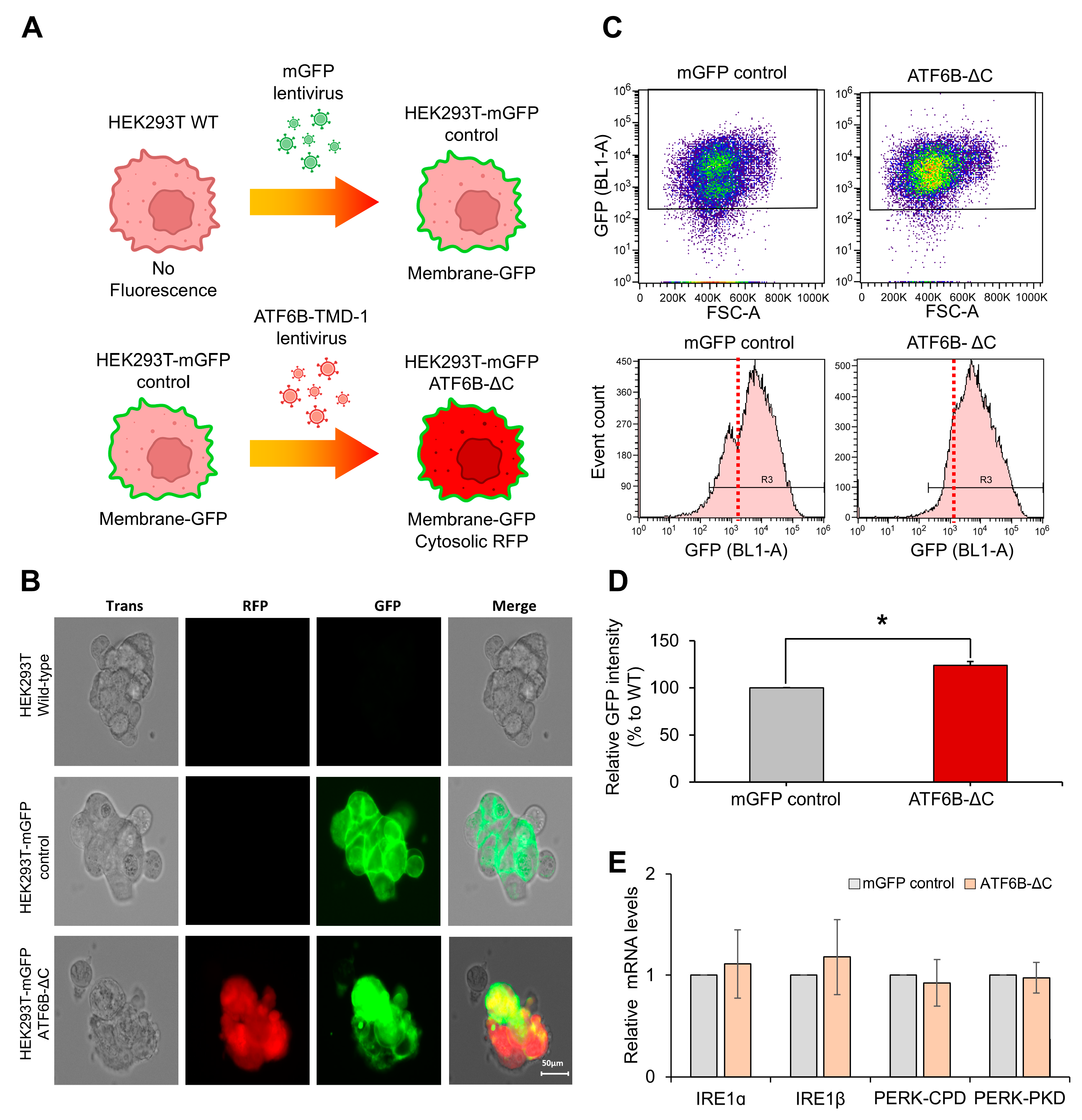
Lentiviral vectors were utilized to create HEK293T cell lines stably expressing Cas9 or mGFP. Lentiviruses containing either the pLV-Cas9.SFFV.Neo or the pLVX-CMV.mGFP.IRES.Puro vectors were transfected into HEK293T cells for this purpose. Selection of stable cell lines expressing Cas9 was achieved using G418 (1000 μg/mL; InvivoGen, San Diego, CA, USA), while those expressing mGFP were selected using Puromycin (2 μg/mL; InvivoGen) over a period of two weeks.
Lentiviral vectors were also utilized to create genome-edited cell lines to knock out the ATF6B gene by CRISPR-Cas9 technology. Briefly, HEK293T cells were transfected with lentiviruses carrying pL-sgRNA.EFS.Zeo.tRFP vectors, each containing a designed sgRNA targeting specific genes. Following the transduction process, the HEK293T cells infected with sgRNAs underwent a rigorous selection phase. This phase involved the application of Zeocin at a concentration of 200 μg/mL (InvivoGen) as the selective agent. This selection process was carried out over a period of two weeks, ensuring that only the cells successfully incorporating the sgRNA remained and proliferated.
2.6. Next-Generation Sequencing (NGS)
In total, 5 × 10
6 cells were harvested, and the genomic DNA of each sample was purified. Genomic DNA purification was performed according to the manufacturer’s instructions. To obtain the amplified ATF6B gene PCR product for each sample, the ATF6B target region was amplified through PCR using the designed primer pair (
Table 2). The first step of PCR conditions was 1 cycle of 5 min at 95 °C, followed by 10 cycles of 10 s at 95 °C, 10 s at 70 °C or 60 °C (annealing temperature was decreased by 1 °C per cycle), and 30 s at 72 °C. Next, the second step of PCR conditions was 30 cycles of 10 s at 95 °C, 10 s at 60 °C, and 30 s at 72 °C, finishing with 3 min incubation at 72 °C.
PCR products were separated on a 1.5% agarose gel in TAE buffer containing 1× RedSafe Nucleic Acid Staining Solution (iNtRon Biotechnology) by electrophoresis and visualized under a Blue LED light. After visualization of the PCR products, the gel was cut, and the PCR band that matched the expected size was collected. Collected PCR products were extracted using the Labopass GEL EXTRACTION KIT (Cosmogenetech, Seoul, Republic of Korea). Gel extraction was performed according to the manufacturer’s instructions. Extracted PCR products were analyzed through an NGS analysis company (Bionics, Seoul, Republic of Korea).
To assess potential off-target effects of the ATF6B sgRNA, in silico prediction was conducted using the Cas-OFFinder tool (
http://www.rgenome.net/cas-offinder/, accessed on 4 February 2025) under mismatch conditions of 1–3 nucleotides. A total of 17 candidate off-target sites were identified. Genomic DNA was extracted, and PCR amplification was performed to generate 900–1200 bp products spanning each predicted site. The amplified products were subjected to next-generation sequencing (BitSeq, Bionics, Seoul, Republic of Korea). The resulting sequence data were analyzed using the Cas-Analyzer tool (
http://www.rgenome.net/cas-analyzer/, accessed on 3 April 2025) to detect indel events. No insertions or deletions were observed at any of the predicted off-target sites.
2.7. Determination of Membranous Protein Yields
For the extraction of membrane proteins, 5 × 106 cells from both ATF6B-ΔC and wild-type HEK293T cell lines were collected. The membrane protein extraction was performed using Mem-PER Plus Membrane Protein Extraction Kit (Thermo Fisher, Inc., Waltham, MA, USA) according to the manufacturer’s instructions. The harvested cells were washed 2 times with Cell Wash Solution (info) and permeabilized with 0.75 mL of Permeabilization Buffer (info) (incubation at 4 °C for 10 min with mixing). After incubation, the samples were centrifuged for 15 min (16,000× g at 4 °C) to separate the supernatant and pellet. The supernatant was discarded, and the pellet was resuspended in 0.5 mL of Solubilization Buffer (info). Then, the resuspended samples were incubated for 30 min at 4 °C with mixing. After that, the mixed samples were separated into supernatant and pellet through centrifugation (15 min/16,000× g/4 °C). Finally, the membrane protein samples (supernatant) were obtained from the centrifugates. The Bradford assay was employed to quantify the production yield of membrane proteins. A total of 20 µL of the extracted membrane proteins was diluted by 180 µL of D.W, and 10 µL of the diluted samples were mixed with 200 µL of 1× Bradford reagent (Bio-Rad Protein Assay Dye Reagent Concentrate; Bio-Rad, Hercules, CA, USA). After 5 min incubation, the absorbance was measured at 595 nm using a photometer (SpectraMax® ABS; Molecular Devices, San Jose, CA, USA).
2.8. Flow Cytometric Analysis
To measure the mGFP intensity of ATF6B-ΔC mGFP-stable HEK293T cells, ATF6B-ΔC and WT mGFP-stable HEK293T cells were seeded into 100 mm cell culture dish prior to the experiment. Cells were cultured to approximately 100% confluency and then harvested by trypsinization. Harvested cells were washed twice with PBS and subsequently resuspended in PBS. Resuspended cells were transferred to new FACS tubes, and GFP intensity was measured through an Attune NxT flow cytometer (Thermo Fisher, Inc.).
2.9. Quantitative Real-Time PCR for UPR-Related Gene Expression
To evaluate the impact of ATF6B deletion on other UPR pathway components, total RNA was extracted from wild-type and ATF6B-ΔC HEK293T cells using TRIzol reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA, USA) on a StepOnePlus™ Real-Time PCR System (Applied Biosystems). The relative expression levels of IRE1α, IRE1β, PERK-CPD, and PERK-PKD were determined using the ΔΔCt method, with GAPDH as the internal control. The following primers were used: IRE1α (forward: 5′-AGGAGGCTGAGGAAGGAGAA-3′, reverse: 5′-TGGTAGGAGGCAGTGAGGAT-3′), IRE1β (forward: 5′-ACGAGGAGAAGGAGGAGATG-3′, reverse: 5′-CTGTGGCAGTAGTAGGAGGA-3′), PERK-CPD (forward: 5′-GGAAGTGTTCCAGCTTCAGG-3′, reverse: 5′-GTCATCAGGCAGCTTTCTCA-3′), PERK-PKD (forward: 5′-AGATGTGGCTGGTGATGTTG-3′, reverse: 5′-TGGCTGAAGATGAGGTTGAA-3′), and GAPDH (forward: 5′-GAAGGTGAAGGTCGGAGT-3′, reverse: 5′-GAAGATGGTGATGGGATTTC-3′).
2.10. Statistical Analysis
The data are expressed as means ± SEM. An unpaired t-test was utilized to compare two groups, considering p < 0.05 as the threshold for statistical significance. The analyses were derived from three independent experimental replicates.
4. Discussion
This study aimed to enhance membrane protein production in HEK293T cells by using CRISPR-Cas9 technology to edit the ATF6B gene. The experimental approach involved optimizing sgRNA efficiency, validating gene editing, and comparing membrane protein production levels between edited and wild-type cells. The sgRNAs targeting ATF6A-TMD, ATF6B-ΔC, IRE1A-KD, IRE1B-KO, PERK-CPD, and PERK-PKD were designed, with ATF6B-ΔC showing the highest efficiency in the T7E1 assay. Sequencing of the ATF6B-ΔC edited cells revealed predominant 11, 14, 1, and 10 bp deletions, which disrupted exon sequences, caused exon skipping, and introduced premature stop codons, leading to suppression of normal protein expression. Comparative analysis of membrane protein production showed a significant increase of approximately 40 ± 17.6% in ATF6B-ΔC cells compared to wild-type cells. Quantitative analysis using PAGE gel and flow cytometry confirmed the enhanced production, with a 23.9 ± 4.2% increase in GFP intensity in ATF6B-ΔC cells.
To investigate the potential off-target effects of ATF6B sgRNA, we used the Cas-OFFinder tool (
http://www.rgenome.net/cas-offinder/) to identify 17 candidate sites under 1–3 bp mismatch conditions. PCR amplicons (900–1200 bp) covering these regions were subjected to NGS (BitSeq, Bionics; Seoul, Republic of Korea), and indel analysis was performed using Cas-Analyzer. No indel mutations were detected at any of the 17 predicted sites, suggesting high specificity of the sgRNA used. A detailed list of the predicted off-target sites and their sequencing results is presented in
Supplementary Table S3. These results indicate that the deletion of ATF6B’s C-terminal domain significantly enhances membrane protein productivity by upregulating the UPR pathway and increasing the cell’s capacity to handle misfolded proteins. This novel approach could have significant implications for the biotechnology and pharmaceutical industries, where membrane proteins are critical targets for drug development and other applications.
Despite the technical challenges associated with membrane protein purification, even moderate improvements in yield can have a profound industrial impact. Membrane proteins are inherently difficult to purify and stabilize due to their hydrophobic nature and structural complexity. However, when such proteins are used as raw materials for biopharmaceutical development, an increase of approximately 20% in production—as demonstrated in this study—represents more than a simple numerical improvement. It directly contributes to lowering manufacturing costs, improving production efficiency, and enhancing scalability. These benefits are particularly critical in the development of high-value or rare membrane protein-based therapeutics, where production bottlenecks often limit progress. Enhancing yield in this context strengthens commercial viability and supports broader accessibility to innovative therapies.
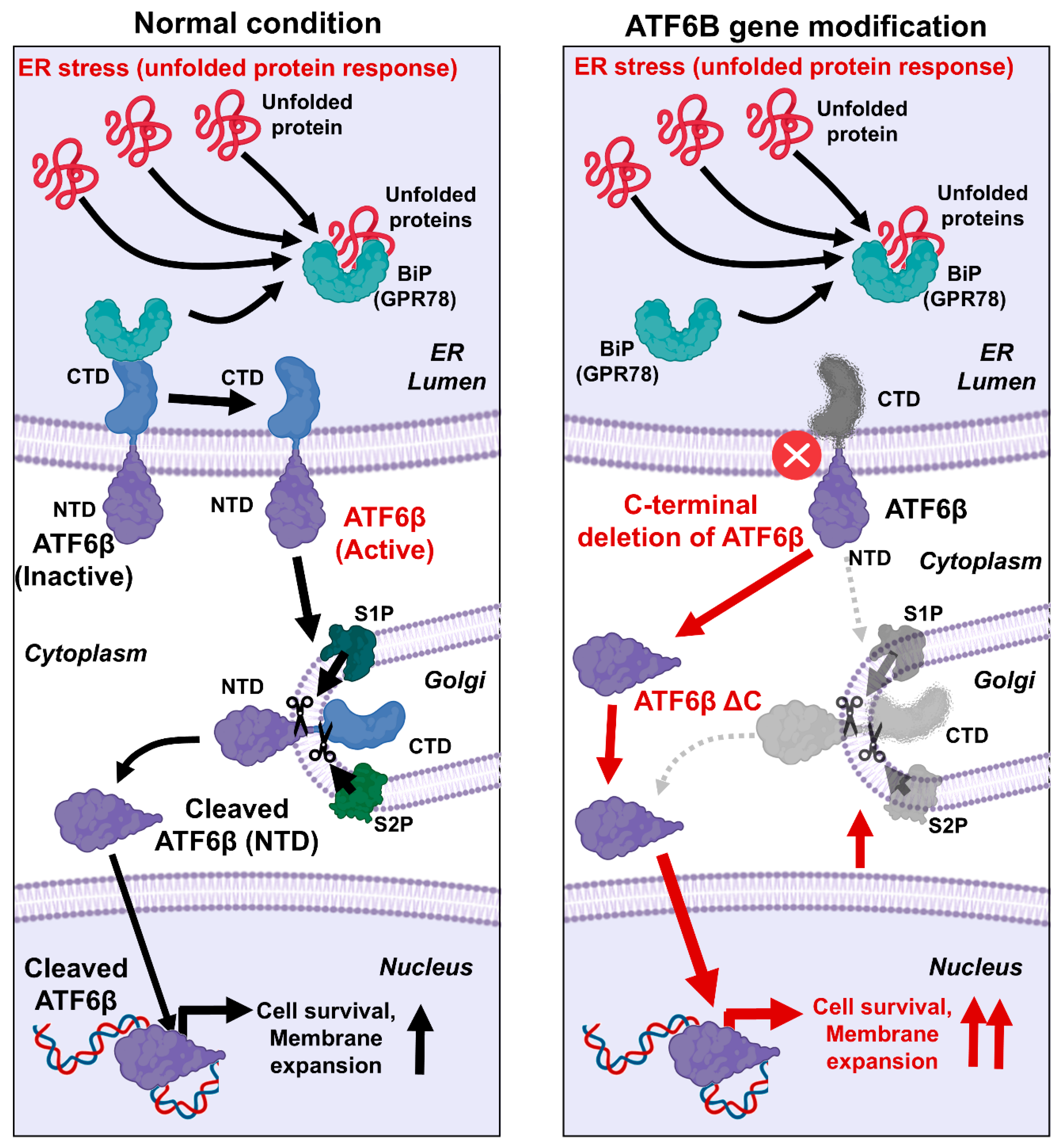
The possible mechanism by which the C-terminal deletion of ATF6B enhances membrane protein productivity is as follows (
Figure 7). The UPR is a crucial cellular process that mitigates the effects of ER stress by enhancing the production of molecular chaperones, degrading misfolded proteins, and attenuating protein synthesis [
1,
2,
3]. The UPR is primarily regulated by three ER membrane-associated proteins: ATF6, PERK, and IRE1. ATF6, upon sensing ER stress, is transported to the Golgi apparatus, where it undergoes proteolytic cleavage by the Site-1 and Site-2 proteases (S1P and S2P) [
1,
3]. This cleavage releases the N-terminal cytoplasmic domain of ATF6, which then translocates to the nucleus to function as a transcription factor. This domain upregulates UPR target genes, including those encoding chaperones and components of the ER-associated degradation pathway. In this research, the C-terminal domain of ATF6B was specifically targeted using CRISPR-Cas9 technology. By deleting this region, it was hypothesized that the N-terminal cytoplasmic domain of ATF6B would be more readily available to enter the nucleus and exert its transcriptional effects. The results confirmed our hypothesis, showing a significant increase in membrane protein production in the ATF6B-ΔC cells compared to wild-type cells. This enhancement can be attributed to the intensified UPR, which maintains ER homeostasis and increases the cell’s capacity to handle misfolded proteins, thereby facilitating greater membrane protein synthesis [
25,
26]. The successful modification of ATF6B-ΔC led to the upregulation of UPR target genes, thereby enhancing the ER’s protein folding and processing capacity. This not only underscores the importance of ATF6B in the UPR pathway but also demonstrates a novel approach to augmenting membrane protein production in HEK293T cells. The successful deletion of the ATF6B C-terminal domain resulted in the upregulation of UPR target genes, enhancing the ER’s protein folding and processing capacity. This finding highlights the critical role of ATF6B in the UPR pathway and presents a novel strategy to boost membrane protein production in HEK293T cells.
The specific deletions in the ATF6B gene, including 11, 14, 1, and 10 bp deletions, were observed in this study as a result of the precise cutting action of the CRISPR-Cas9 system. These deletions led to the disruption of exon sequences, causing exon skipping and the introduction of premature stop codons. The CRISPR-Cas9 system was designed with sgRNAs targeting specific regions of the ATF6B gene, which directed the Cas9 nuclease to create double-strand breaks at these sites. During the repair process, the non-homologous end joining (NHEJ) pathway, which often results in insertions or deletions, repaired the breaks inaccurately, leading to the observed deletions. These genetic alterations disrupted the normal reading frame of the ATF6B gene, producing truncated, non-functional proteins or leading to the complete absence of ATF6B protein due to nonsense-mediated decay. Consequently, the disruption of the ATF6B gene by these deletions could lead to enhanced upregulation of UPR target genes and improved ER protein folding and processing capacity. This result highlights the critical role of ATF6B in the UPR pathway and presents a novel strategy to boost membrane protein production in HEK293T cells. Notably, despite the loss of ATF6B function, no significant reduction in cell viability was observed, suggesting that compensatory ER stress pathways may have helped preserve cell survival.
While the enhancement of membrane protein production via ATF6B editing holds significant promise for drug discovery and biomanufacturing, translating this approach to industrial-scale applications presents several challenges. One major concern is the long-term stability of the edited cell line. Gene editing, particularly when targeting stress response pathways like ATF6, may lead to subtle changes in cell growth behavior or genomic stability over extended culture periods, which could reduce productivity over time. Moreover, applying this system in large-scale bioreactor environments requires careful optimization, as cellular responses may vary under different culture conditions. These aspects highlight the need for further development before this platform can be widely adopted in industrial bioprocesses.
This study has several limitations. Although the C-terminal deletion of ATF6B significantly improved membrane protein production, the long-term stability and viability of the edited cells under large-scale production conditions remain untested. Potential off-target effects of CRISPR-Cas9 editing were not extensively evaluated, and the study focused solely on ATF6B, leaving other potentially synergistic genes unexplored. Additionally, the use of GFP as a reporter limits generalizability to other membrane proteins. Despite these limitations, this study offers a valuable proof-of-concept for using CRISPR-Cas9 to enhance membrane protein production and presents a novel strategy for modulating the UPR pathway to improve bioproduction efficiency.
In a broader context, the development of gene-edited cell lines for enhanced protein production also aligns with global efforts to address sustainability and food security challenges. As highlighted by Saleem et al. [
28], biotechnology plays an essential role in achieving the Sustainable Development Goals (SDGs) by improving resource efficiency and supporting resilient systems under climate pressure. Our approach, while focused on biopharmaceutical production, may contribute to these broader goals by promoting more efficient and scalable biologics manufacturing platforms. In summary, this study demonstrates that CRISPR-Cas9-mediated deletion of the ATF6B transmembrane domain significantly enhances membrane protein production in HEK293T cells. Optimizing sgRNA efficiency and validating gene editing revealed that ATF6B-ΔC cells exhibited deletions that disrupted exon sequences and introduced premature stop codons, effectively suppressing normal protein expression. The gene editing resulted in a substantial increase in membrane protein production, highlighting the potential of targeting the ATF6B gene to upregulate the UPR pathway and improve cellular capacity for managing misfolded proteins. This approach holds promise for advancing biotechnology and pharmaceutical applications where efficient membrane protein production is critical.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}