

Mesenchymal Stem Cell-Conditioned Media-Loaded Microparticles Enhance Acute Patency in Silk-Based Vascular Grafts

 , , ,
, , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
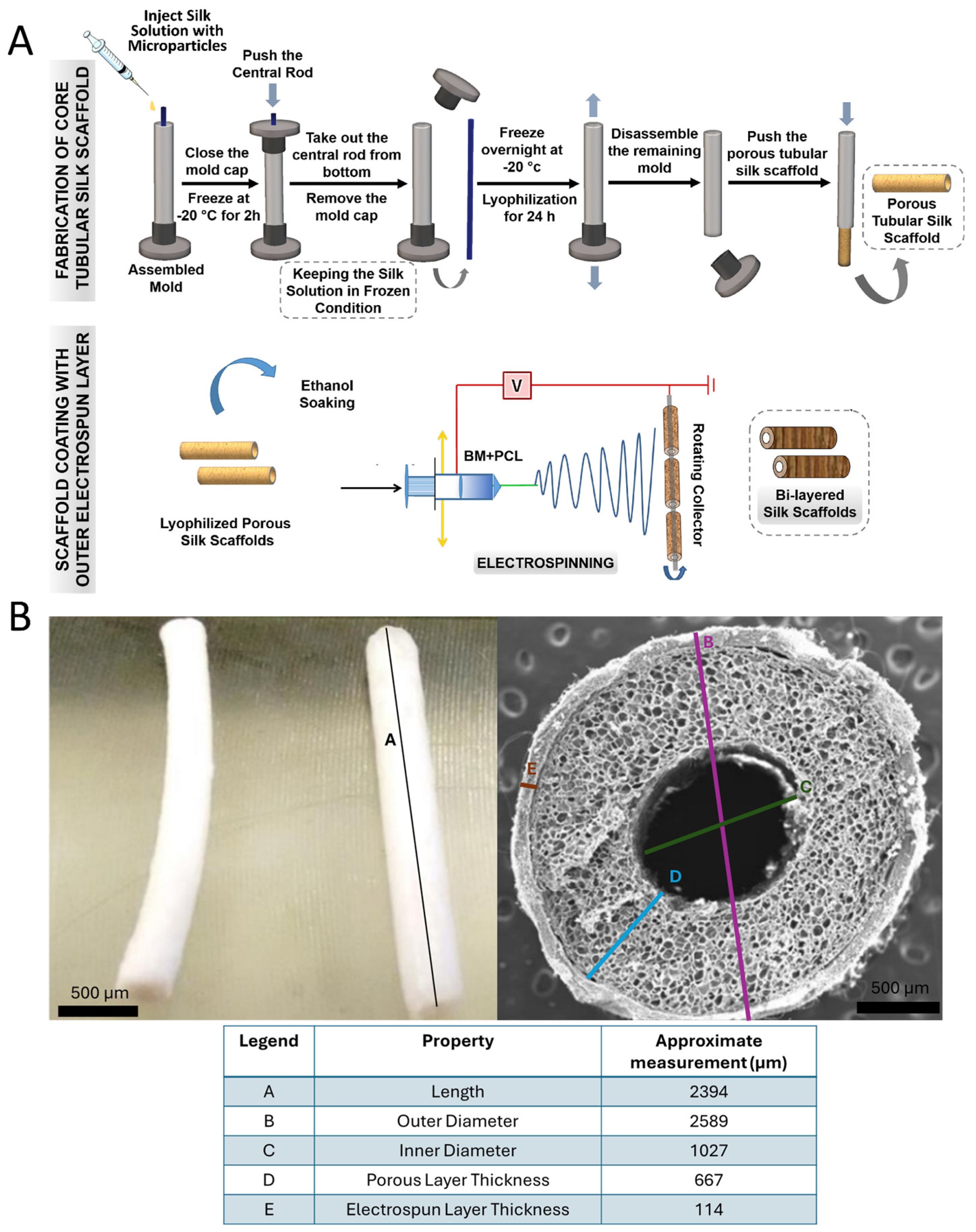
2. Materials and Methods
2.1. Conditioned Media (CM) Generation
2.2. CM Microparticle (ArtMSC) Fabrication
2.3. Characterization of CM and ArtMSC Morphology and Release
2.4. Vascular Cell Culture
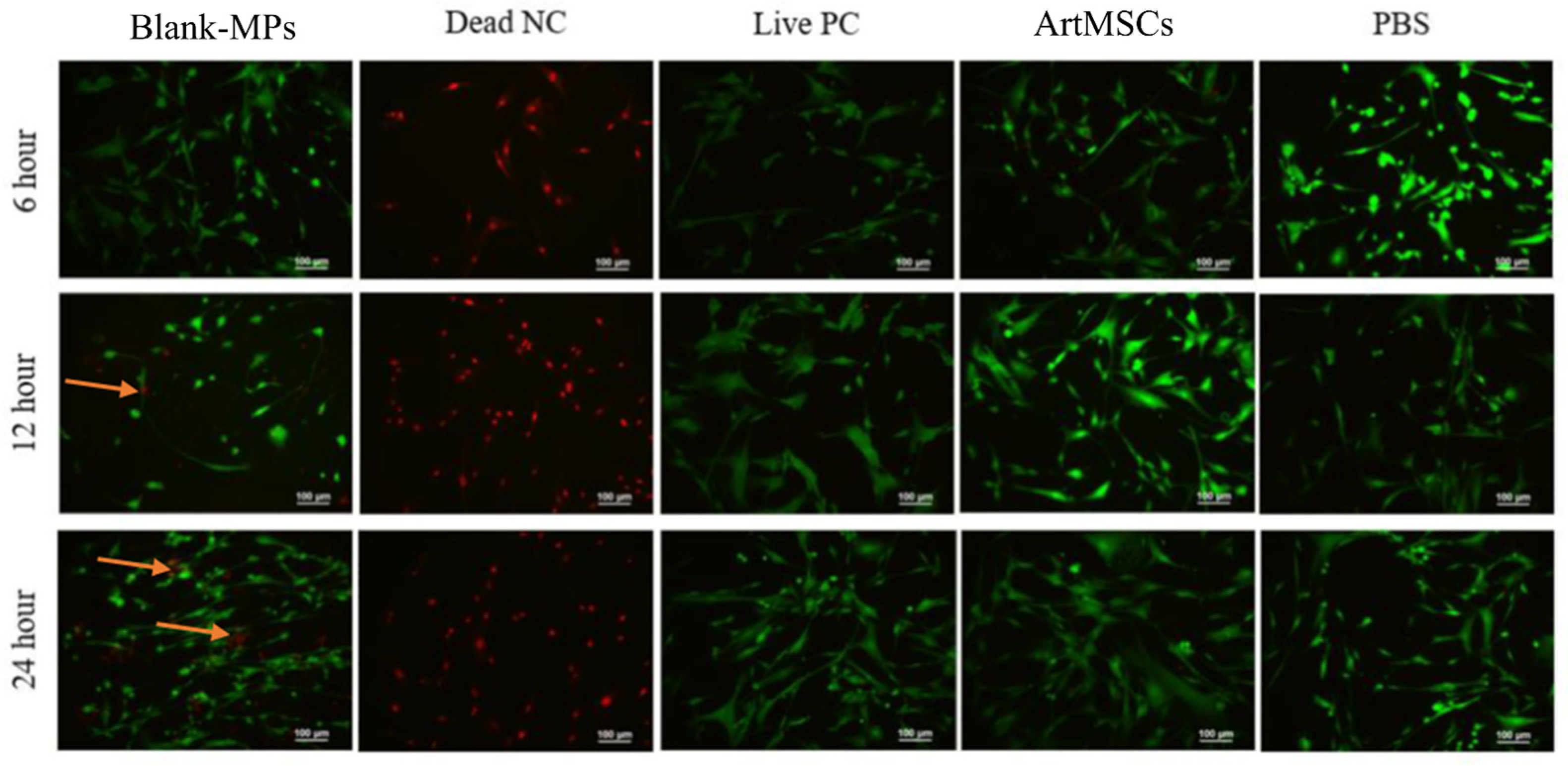
2.5. MP Toxicity Assessment
2.6. SMC and EC Proliferation
2.7. Fabrication of MP Lyogel Silk Constructs
2.8. In Vivo Implantation of ArtMSC Lyogel Silk Constructs
2.9. Immunofluorescent Staining and Histochemical Analysis
2.10. Statistical Analysis
3. Results
3.1. Characterization of ArtMSCs
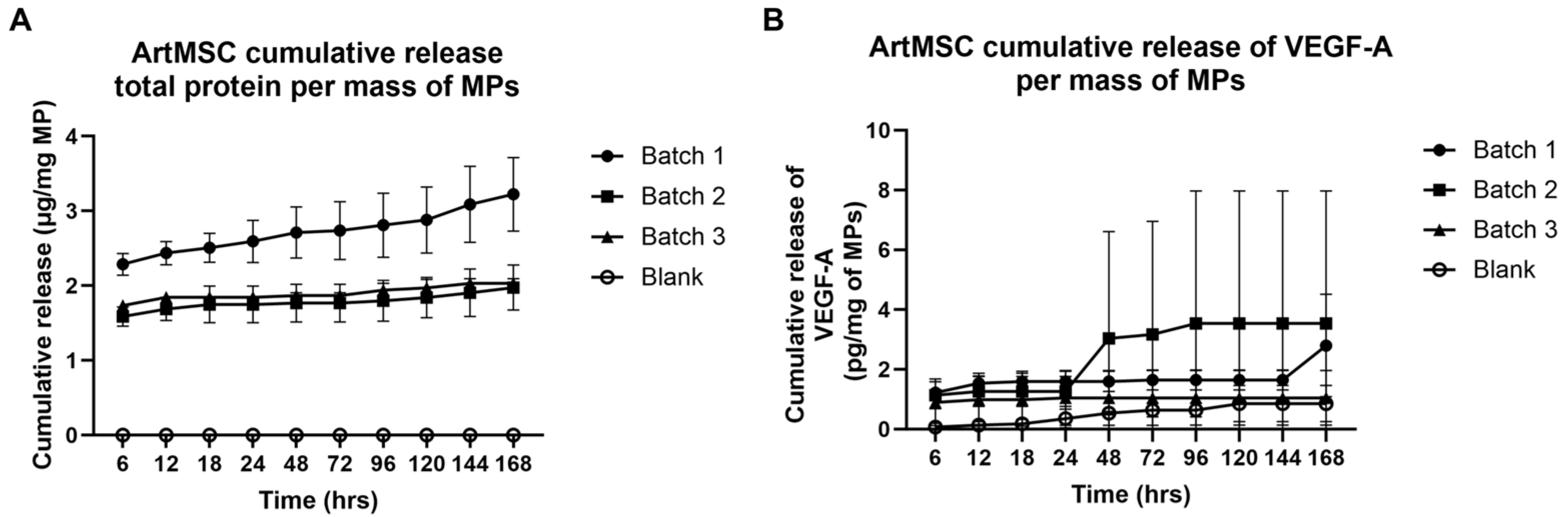
3.2. Protein Release from ArtMSCs
3.3. In Vitro Analysis of CM and ArtMSCs
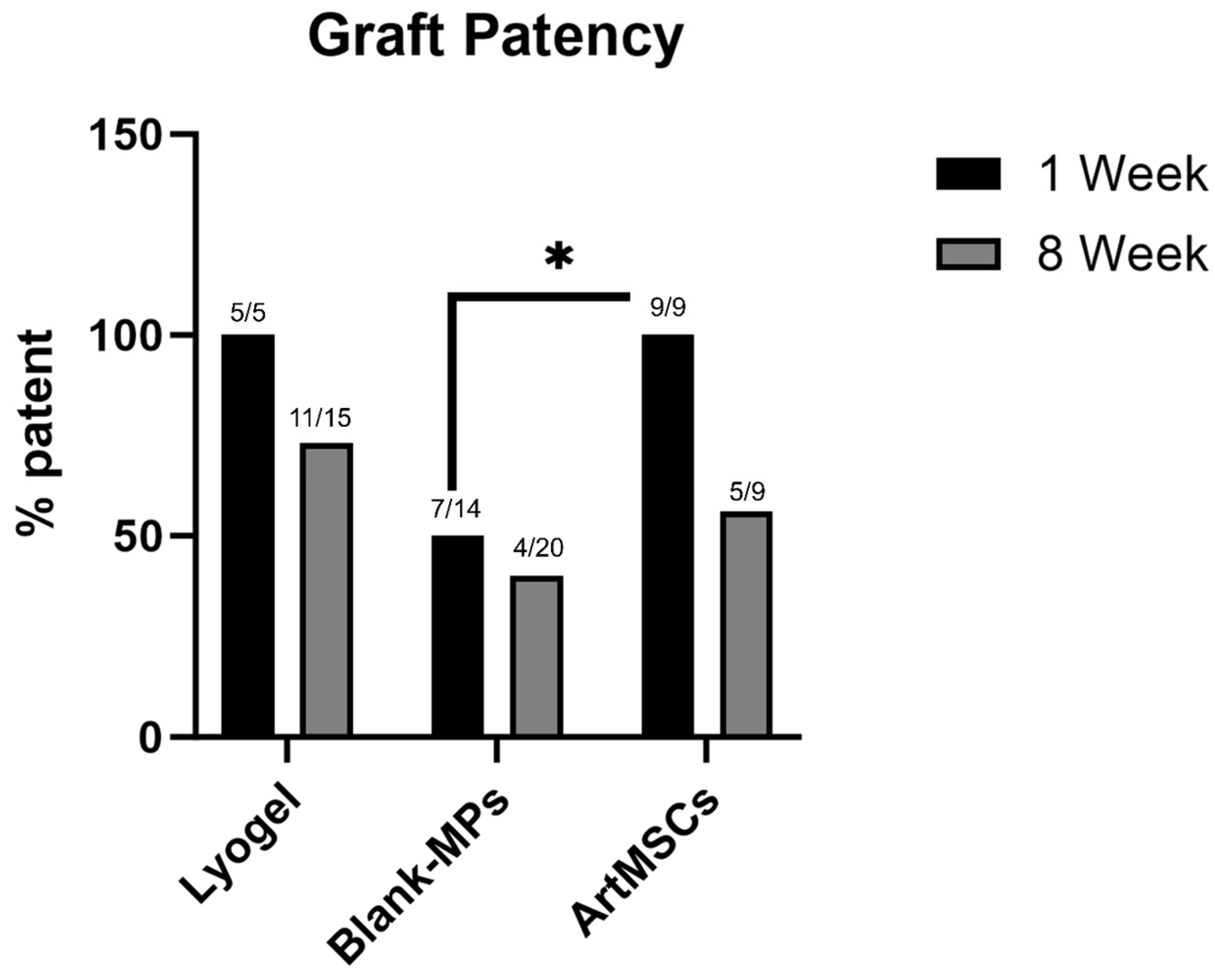
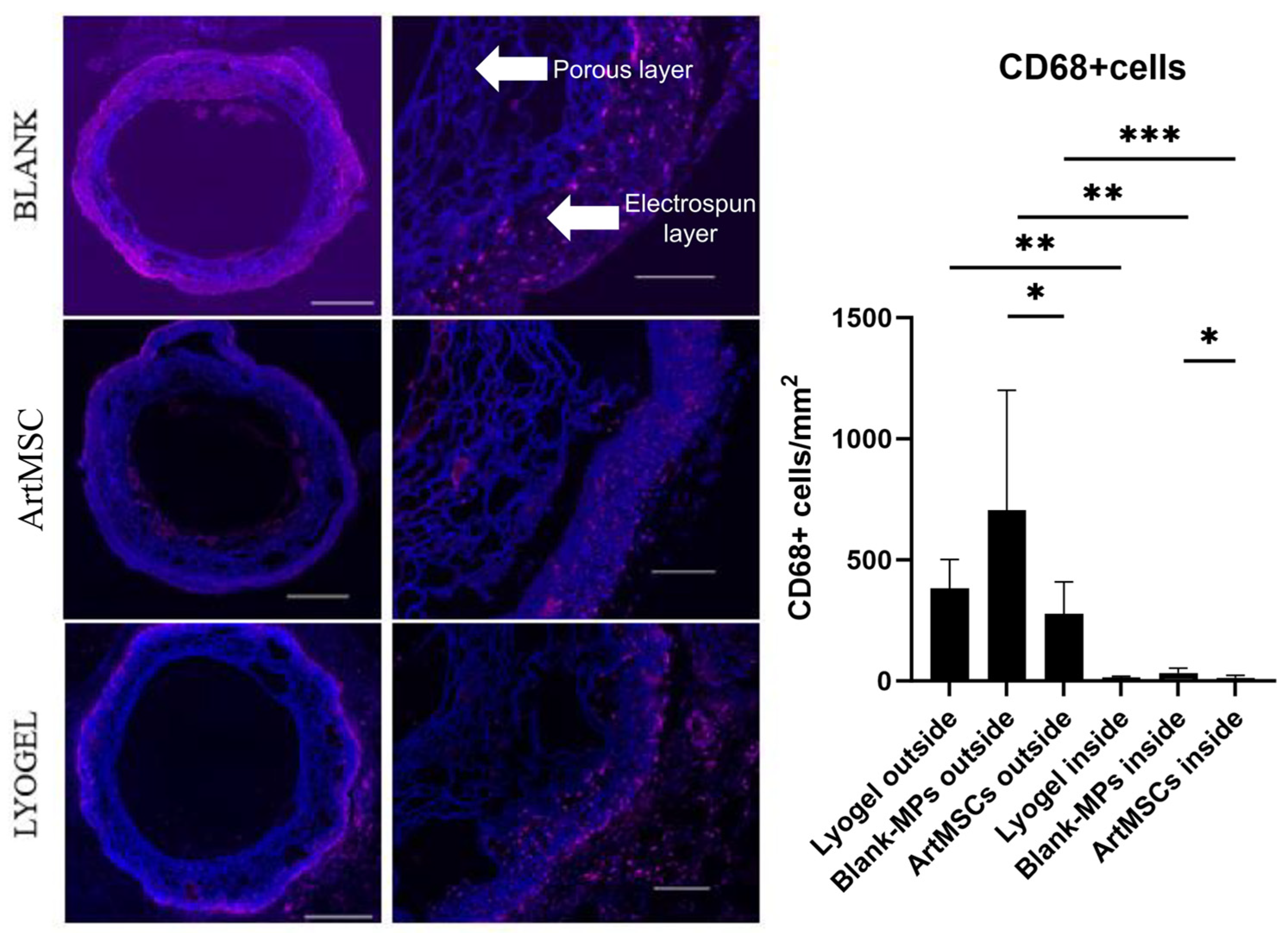
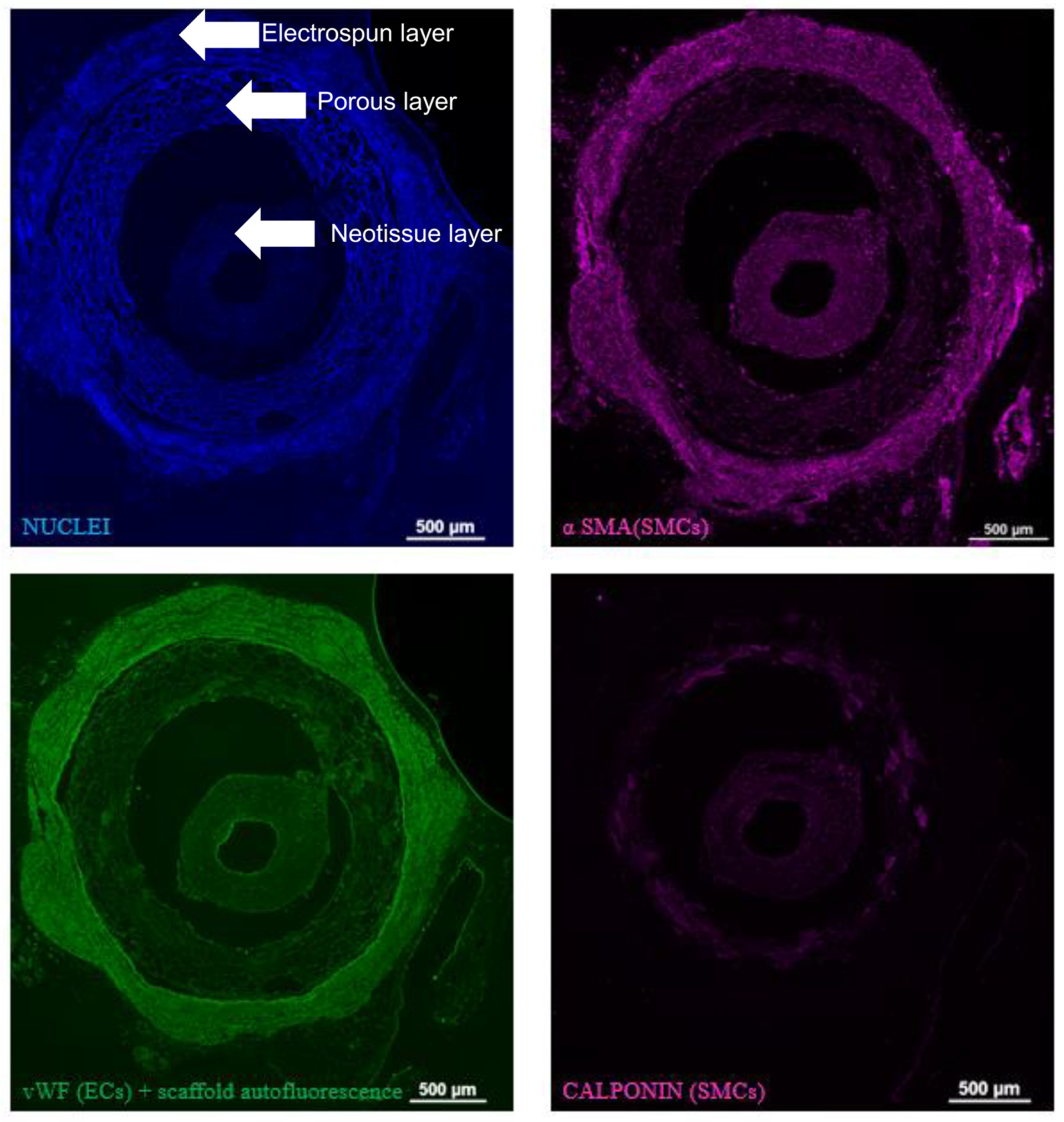
3.4. Explant Analysis of ArtMSC Lyogel TEVGs
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heart Disease Facts. 2024. Available online: https://www.cdc.gov/heart-disease/data-research/facts-stats/index.html (accessed on 10 September 2024).
- Cardiovascular Diseases (CVDs). 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)#:~:text=An%20estimated%2017.9%20million%20people,to%20heart%20attack%20and%20stroke (accessed on 10 September 2024).
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [PubMed]
- Harskamp, R.E.; Alexander, J.H.; Ferguson, T.B.; Hager, R.; Mack, M.J.; Englum, B.; Wojdyla, D.; Schulte, P.J.; Kouchoukos, N.T.; De Winter, R.J.; et al. Frequency and Predictors of Internal Mammary Artery Graft Failure and Subsequent Clinical Outcomes. Circulation 2016, 133, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Xenogiannis, I.; Zenati, M.; Bhatt, D.L.; Rao, S.V.; Rodés-Cabau, J.; Goldman, S.; Shunk, K.A.; Mavromatis, K.; Banerjee, S.; Alaswad, K.; et al. Saphenous Vein Graft Failure: From Pathophysiology to Prevention and Treatment Strategies. Circulation 2021, 144, 728–745. [Google Scholar] [CrossRef]
- Cunnane, E.M.; Lorentz, K.L.; Ramaswamy, A.K.; Gupta, P.; Mandal, B.B.; O’Brien, F.J.; Weinbaum, J.S.; Vorp, D.A. Extracellular Vesicles Enhance the Remodeling of Cell-Free Silk Vascular Scaffolds in Rat Aortae. ACS Appl. Mater. Interfaces 2020, 12, 26955–26965. [Google Scholar] [CrossRef]
- Gupta, P.; Lorentz, K.L.; Haskett, D.G.; Cunnane, E.M.; Ramaswamy, A.K.; Weinbaum, J.S.; Vorp, D.A.; Mandal, B.B. Bioresorbable silk grafts for small diameter vascular tissue engineering applications: In vitro and in vivo functional analysis. Acta Biomater. 2020, 105, 146–158. [Google Scholar] [CrossRef]
- Haskett, D.G.; Saleh, K.S.; Lorentz, K.L.; Josowitz, A.D.; Luketich, S.K.; Weinbaum, J.S.; Kokai, L.E.; D’Amore, A.; Marra, K.G.; Rubin, J.P.; et al. An exploratory study on the preparation and evaluation of a “same-day” adipose stem cell-based tissue-engineered vascular graft. J. Thorac. Cardiovasc. Surg. 2018, 156, 1814–1822.e3. [Google Scholar] [CrossRef]
- Krawiec, J.T.; Weinbaum, J.S.; Liao, H.T.; Ramaswamy, A.K.; Pezzone, D.J.; Josowitz, A.D.; D’Amore, A.; Rubin, J.P.; Wagner, W.R.; Vorp, D.A. In Vivo Functional Evaluation of Tissue-Engineered Vascular Grafts Fabricated Using Human Adipose-Derived Stem Cells from High Cardiovascular Risk Populations. Tissue Eng. Part A 2016, 22, 765–775. [Google Scholar] [CrossRef]
- Lorentz, K.L.; Gupta, P.; Shehabeldin, M.S.; Cunnane, E.M.; Ramaswamy, A.K.; Verdelis, K.; DiLeo, M.V.; Little, S.R.; Weinbaum, J.S.; Sfeir, C.S.; et al. CCL2 loaded microparticles promote acute patency in silk-based vascular grafts implanted in rat aortae. Acta Biomater. 2021, 135, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Krawiec, J.T.; Liao, H.T.; Kwan, L.L.; D’Amore, A.; Weinbaum, J.S.; Rubin, J.P.; Wagner, W.R.; Vorp, D.A. Evaluation of the stromal vascular fraction of adipose tissue as the basis for a stem cell-based tissue-engineered vascular graft. J. Vasc. Surg. 2017, 66, 883–890.e1. [Google Scholar] [CrossRef]
- L’Heureux, N.; Dusserre, N.; Konig, G.; Victor, B.; Keire, P.; Wight, T.N.; Chronos, N.A.; Kyles, A.E.; Gregory, C.R.; Hoyt, G.; et al. Human tissue-engineered blood vessels for adult arterial revascularization. Nat. Med. 2006, 12, 361–365. [Google Scholar] [CrossRef]
- Syedain, Z.; Reimer, J.; Lahti, M.; Berry, J.; Johnson, S.; Tranquillo, R.T. Tissue engineering of acellular vascular grafts capable of somatic growth in young lambs. Nat. Commun. 2016, 7, 12951. [Google Scholar] [CrossRef] [PubMed]
- Hashi, C.K.; Zhu, Y.; Yang, G.-Y.; Young, W.L.; Hsiao, B.S.; Wang, K.; Chu, B.; Li, S. Antithrombogenic property of bone marrow mesenchymal stem cells in nanofibrous vascular grafts. Proc. Natl. Acad. Sci. USA 2007, 104, 11915–11920. [Google Scholar] [CrossRef]
- Luo, J.; Qin, L.; Zhao, L.; Gui, L.; Ellis, M.W.; Huang, Y.; Kural, M.H.; Clark, J.A.; Ono, S.; Wang, J.; et al. Tissue-Engineered Vascular Grafts with Advanced Mechanical Strength from Human iPSCs. Cell Stem Cell 2020, 26, 251–261.e8. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, C.B.; Bell, E. A Blood Vessel Model Constructed from Collagen and Cultured Vascular Cells. Science 1986, 231, 397–400. [Google Scholar] [CrossRef] [PubMed]
- L’Heureux, N.; Pâquet, S.; Labbé, R.; Germain, L.; Auger, F.A. A completely biological tissue-engineered human blood vessel. FASEB J. 1998, 12, 47–56. [Google Scholar] [PubMed]
- Niklason, L.E.; Gao, J.; Abbott, W.M.; Hirschi, K.K.; Houser, S.; Marini, R.; Langer, R. Functional Arteries Grown in Vitro. Science 1999, 284, 489–493. [Google Scholar] [CrossRef]
- Gupta, P.; Mandal, B.B. Tissue-Engineered Vascular Grafts: Emerging Trends and Technologies. Adv. Funct. Mater. 2021, 31, 2100027. [Google Scholar] [CrossRef]
- Hibino, N.; McGillicuddy, E.; Matsumura, G.; Ichihara, Y.; Naito, Y.; Breuer, C.; Shinoka, T. Late-term results of tissue-engineered vascular grafts in humans. J. Thorac. Cardiovasc. Surg. 2010, 139, 431–436.e2. [Google Scholar] [CrossRef]
- Drews, J.D.; Pepper, V.K.; Best, C.A.; Szafron, J.M.; Cheatham, J.P.; Yates, A.R.; Hor, K.N.; Zbinden, J.C.; Chang, Y.C.; Mirhaidari, G.J.M.; et al. Spontaneous reversal of stenosis in tissue-engineered vascular grafts. Sci. Transl. Med. 2020, 12, eaax6919. [Google Scholar] [CrossRef]
- Smith, R.J.; Nasiri, B.; Kann, J.; Yergeau, D.; Bard, J.E.; Swartz, D.D.; Andreadis, S.T. Endothelialization of arterial vascular grafts by circulating monocytes. Nat. Commun. 2020, 11, 1622. [Google Scholar] [CrossRef]
- Nasiri, B.; Row, S.; Smith, R.J., Jr.; Swartz, D.D.; Andreadis, S.T. Cell-free vascular grafts that grow with the host. Adv. Funct. Mater. 2020, 30, 202005769. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Yi, T.; Nasiri, B.; Breuer, C.K.; Andreadis, S.T. Implantation of VEGF-functionalized cell-free vascular grafts: Regenerative and immunological response. FASEB J. 2019, 33, 5089–5100. [Google Scholar] [CrossRef] [PubMed]
- Pashneh-Tala, S.; MacNeil, S.; Claeyssens, F. The Tissue-Engineered Vascular Graft-Past, Present, and Future. Tissue Eng. Part B Rev. 2016, 22, 68–100. [Google Scholar] [CrossRef]
- Gupta, P.; Mandal, B.B. Silk biomaterials for vascular tissue engineering applications. Acta Biomater. 2021, 134, 79–106. [Google Scholar] [CrossRef]
- Janani, G.; Kumar, M.; Chouhan, D.; Moses, J.C.; Gangrade, A.; Bhattacharjee, S.; Mandal, B.B. Insight into Silk-Based Biomaterials: From Physicochemical Attributes to Recent Biomedical Applications. ACS Appl. Bio Mater. 2019, 2, 5460–5491. [Google Scholar] [CrossRef]
- Matsuda, K.; Falkenberg, K.J.; Woods, A.A.; Choi, Y.S.; Morrison, W.A.; Dilley, R.J. Adipose-Derived Stem Cells Promote Angiogenesis and Tissue Formation for In Vivo Tissue Engineering. Tissue Eng. Part A 2013, 19, 1327–1335. [Google Scholar] [CrossRef]
- Cun, X.; Hosta-Rigau, L. Topography: A Biophysical Approach to Direct the Fate of Mesenchymal Stem Cells in Tissue Engineering Applications. Nanomaterials 2020, 10, 2070. [Google Scholar] [CrossRef]
- Ellis, B.W.; Traktuev, D.O.; Merfeld-Clauss, S.; Can, U.I.; Wang, M.; Bergeron, R.; Zorlutuna, P.; March, K.L. Adipose stem cell secretome markedly improves rodent heart and human induced pluripotent stem cell-derived cardiomyocyte recovery from cardioplegic transport solution exposure. Stem Cells 2021, 39, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, T.; Sun, J.; Shen, M.; Xue, X.; Chen, Y.; Zhang, Z. Harnessing the secretome of adipose-derived stem cells in the treatment of ischemic heart diseases. Stem Cell Res. Ther. 2019, 10, 196. [Google Scholar] [CrossRef]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of Angiogenic and Antiapoptotic Factors by Human Adipose Stromal Cells. Circulation 2004, 109, 1292–1298. [Google Scholar] [CrossRef]
- Bruk, L.A.; Fan, X.; Resnick, J.L.; Dileo, M.V. Controlled Release of Mesenchymal Stem Cell-Conditioned Media from a Microsphere/Gel-Based Drug Delivery System for Wound Healing of Tympanic Membrane Perforations. J. Tissue Eng. Regen. Med. 2023, 2023, 6039254. [Google Scholar] [CrossRef]
- Delarosa, O.; Lombardo, E.; Beraza, A.; Mancheño-Corvo, P.; Ramirez, C.; Menta, R.; Rico, L.; Camarillo, E.; García, L.; Abad, J.L.; et al. Requirement of IFN-γ–Mediated Indoleamine 2,3-Dioxygenase Expression in the Modulation of Lymphocyte Proliferation by Human Adipose–Derived Stem Cells. Tissue Eng. Part A 2009, 15, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, A.K.; Sides, R.E.; Cunnane, E.M.; Lorentz, K.L.; Reines, L.M.; Vorp, D.A.; Weinbaum, J.S. Adipose-derived stromal cell secreted factors induce the elastogenesis cascade within 3D aortic smooth muscle cell constructs. Matrix Biol. Plus 2019, 4, 100014. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, E.M.; Ramaswamy, A.K.; Lorentz, K.L.; Vorp, D.A.; Weinbaum, J.S. Extracellular Vesicles Derived from Primary Adipose Stromal Cells Induce Elastin and Collagen Deposition by Smooth Muscle Cells within 3D Fibrin Gel Culture. Bioengineering 2021, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tong, M.; Hu, S.; Chen, X. The Bioactive Substance Secreted by MSC Retards Mouse Aortic Vascular Smooth Muscle Cells Calcification. BioMed Res. Int. 2018, 2018, 6053567. [Google Scholar] [CrossRef]
- Ceserani, V.; Ferri, A.; Berenzi, A.; Benetti, A.; Ciusani, E.; Pascucci, L.; Bazzucchi, C.; Coccè, V.; Bonomi, A.; Pessina, A.; et al. Angiogenic and anti-inflammatory properties of micro-fragmented fat tissue and its derived mesenchymal stromal cells. Vasc. Cell 2016, 8, 3. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Coleman, M.E. Conditioned Medium of Mesenchymal Stromal Cells: A New Class of Therapeutics. Biochemistry 2019, 84, 1375–1389. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yoshizawa-Smith, S.; Glowacki, A.; Maltos, K.; Pacheco, C.; Shehabeldin, M.; Mulkeen, M.; Myers, N.; Chong, R.; Verdelis, K.; et al. Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models. J. Dent. Res. 2019, 98, 200–208. [Google Scholar] [CrossRef]
- Soletti, L.; Nieponice, A.; Hong, Y.; Ye, S.H.; Stankus, J.J.; Wagner, W.R.; Vorp, D.A. In vivo performance of a phospholipid-coated bioerodable elastomeric graft for small-diameter vascular applications. J. Biomed. Mater. Res. Part A 2011, 96A, 436–448. [Google Scholar] [CrossRef]
- Wulff, B.; Stahlhoff, S.; Vonthein, R.; Schmidt, A.; Sigler, M.; Torsello, G.B.; Herten, M. Biomimetic Heparan Sulfate-like Coated ePTFE Grafts Reduce In-graft Neointimal Hyperplasia in Ovine Carotids. Ann. Vasc. Surg. 2017, 40, 274–284. [Google Scholar] [CrossRef]
- Khait, L.; Birla, R.K. Bypassing the Patient: Comparison of Biocompatible Models for the Future of Vascular Tissue Engineering. Cell Transplant. 2012, 21, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, D.; Zhu, X.; Liu, N.; Zhang, H.; Tang, R.; Liu, Z. Development of a decellularized human amniotic membrane-based electrospun vascular graft capable of rapid remodeling for small-diameter vascular applications. Acta Biomater. 2022, 152, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Friess, W.; Schlapp, M. Release mechanisms from gentamicin loaded poly(lactic-co-glycolic acid) (PLGA) microparticles. J. Pharm. Sci. 2002, 91, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Hibino, N.; Mejias, D.; Pietris, N.; Dean, E.; Yi, T.; Best, C.; Shinoka, T.; Breuer, C. The innate immune system contributes to tissue-engineered vascular graft performance. FASEB J. 2015, 29, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; King, M.W. Immunomodulation Strategies for the Successful Regeneration of a Tissue-Engineered Vascular Graft. Adv. Healthc. Mater. 2022, 11, 2200045. [Google Scholar] [CrossRef]
- Hibino, N.; Yi, T.; Duncan, D.R.; Rathore, A.; Dean, E.; Naito, Y.; Dardik, A.; Kyriakides, T.; Madri, J.; Pober, J.S.; et al. A critical role for macrophages in neovessel formation and the development of stenosis in tissue-engineered vascular grafts. FASEB J. 2011, 25, 4253–4263. [Google Scholar] [CrossRef]
- Brown, B.N.; Ratner, B.D.; Goodman, S.B.; Amar, S.; Badylak, S.F. Macrophage polarization: An opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials 2012, 33, 3792–3802. [Google Scholar] [CrossRef]
- Hachim, D.; Iftikhar, A.; Lopresti, S.T.; Nolfi, A.L.; Ravichandar, S.; Skillen, C.D.; Brown, B.N. Distinct release strategies are required to modulate macrophage phenotype in young versus aged animals. J. Control. Release 2019, 305, 65–74. [Google Scholar] [CrossRef]
- Pineda Molina, C.; Giglio, R.; Gandhi, R.M.; Sicari, B.M.; Londono, R.; Hussey, G.S.; Bartolacci, J.G.; Quijano Luque, L.M.; Cramer, M.C.; Dziki, J.L.; et al. Comparison of the host macrophage response to synthetic and biologic surgical meshes used for ventral hernia repair. J. Immunol. Regen. Med. 2019, 3, 13–25. [Google Scholar] [CrossRef]
- Brown, B.N.; Londono, R.; Tottey, S.; Zhang, L.; Kukla, K.A.; Wolf, M.T.; Daly, K.A.; Reing, J.E.; Badylak, S.F. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012, 8, 978–987. [Google Scholar] [CrossRef]
- Li, L.; Qian, Y.; Lin, C.; Li, H.; Jiang, C.; Lv, Y.; Liu, W.; Cai, K.; Germershaus, O.; Yang, L. The effect of silk gland sericin protein incorporation into electrospun polycaprolactone nanofibers on in vitro and in vivo characteristics. J. Mater. Chem. B 2015, 3, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, D.; Li, L.; Qian, Z.; He, W.; Ding, R.; Liu, H.; Fan, Y. Effect of Electrospun Silk Fibroin–Silk Sericin Films on Macrophage Polarization and Vascularization. ACS Biomater. Sci. Eng. 2020, 6, 3502–3512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cui, Y.; Wang, J.; Yang, X.; Wu, Y.; Wang, K.; Gao, X.; Li, D.; Li, Y.; Zheng, X.L.; et al. The effect of thick fibers and large pores of electrospun poly(epsilon-caprolactone) vascular grafts on macrophage polarization and arterial regeneration. Biomaterials 2014, 35, 5700–5710. [Google Scholar] [CrossRef]
- Roh, J.D.; Sawh-Martinez, R.; Brennan, M.P.; Jay, S.M.; Devine, L.; Rao, D.A.; Yi, T.; Mirensky, T.L.; Nalbandian, A.; Udelsman, B.; et al. Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 4669–4674. [Google Scholar] [CrossRef]
- Chan, A.H.P.; Filipe, E.C.; Tan, R.P.; Santos, M.; Yang, N.; Hung, J.; Feng, J.; Nazir, S.; Benn, A.J.; Ng, M.K.C.; et al. Altered processing enhances the efficacy of small-diameter silk fibroin vascular grafts. Sci. Rep. 2019, 9, 17461. [Google Scholar] [CrossRef]
- Mohammadi, M.; Luong, J.C.; Rodriguez, S.M.; Cao, R.; Wheeler, A.E.; Lau, H.; Li, S.; Shabestari, S.K.; Chadarevian, J.P.; Alexander, M.; et al. Controlled Release of Stem Cell Secretome Attenuates Inflammatory Response against Implanted Biomaterials. Adv. Healthc. Mater. 2020, 9, 1901874. [Google Scholar] [CrossRef]
- Bissell, M.J.; Aggeler, J. Dynamic reciprocity: How do extracellular matrix and hormones direct gene expression? Prog. Clin. Biol. Res. 1987, 249, 251–262. [Google Scholar] [PubMed]
- Perugini, P.; Genta, I.; Conti, B.; Modena, T.; Pavanetto, F. Long-term release of clodronate from biodegradable microspheres. AAPS PharmSciTech 2001, 2, 6–14. [Google Scholar] [CrossRef]
- Bhang, S.H.; Lee, S.; Shin, J.-Y.; Lee, T.-J.; Jang, H.-K.; Kim, B.-S. Efficacious and Clinically Relevant Conditioned Medium of Human Adipose-derived Stem Cells for Therapeutic Angiogenesis. Mol. Ther. 2014, 22, 862–872. [Google Scholar] [CrossRef]
- Binder, B.Y.K.; Sagun, J.E.; Leach, J.K. Reduced Serum and Hypoxic Culture Conditions Enhance the Osteogenic Potential of Human Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2015, 11, 387–393. [Google Scholar] [CrossRef]
- Ho, S.S.; Hung, B.P.; Heyrani, N.; Lee, M.A.; Leach, J.K. Hypoxic Preconditioning of Mesenchymal Stem Cells with Subsequent Spheroid Formation Accelerates Repair of Segmental Bone Defects. Stem Cells 2018, 36, 1393–1403. [Google Scholar] [CrossRef]
- Ferreira, J.R.; Teixeira, G.Q.; Santos, S.G.; Barbosa, M.A.; Almeida-Porada, G.; Gonçalves, R.M. Mesenchymal Stromal Cell Secretome: Influencing Therapeutic Potential by Cellular Pre-conditioning. Front. Immunol. 2018, 9, 2837. [Google Scholar] [CrossRef]
- Miyachi, H.; Reinhardt, J.W.; Otsuru, S.; Tara, S.; Nakayama, H.; Yi, T.; Lee, Y.-U.; Miyamoto, S.; Shoji, T.; Sugiura, T.; et al. Bone marrow-derived mononuclear cell seeded bioresorbable vascular graft improves acute graft patency by inhibiting thrombus formation via platelet adhesion. Int. J. Cardiol. 2018, 266, 61–66. [Google Scholar] [CrossRef]
- Sohling, N.; Ondreka, M.; Kontradowitz, K.; Reichel, T.; Marzi, I.; Henrich, D. Early Immune Response in Foreign Body Reaction Is Implant/Material Specific. Materials 2022, 15, 2195. [Google Scholar] [CrossRef]
- Gu, B.; Papadimitrakopoulos, F.; Burgess, D.J. PLGA microsphere/PVA hydrogel coatings suppress the foreign body reaction for 6 months. J. Control. Release 2018, 289, 35–43. [Google Scholar] [CrossRef]
- Dahan, N.; Sarig, U.; Bronshtein, T.; Baruch, L.; Karram, T.; Hoffman, A.; Machluf, M. Dynamic Autologous Reendothelialization of Small-Caliber Arterial Extracellular Matrix: A Preclinical Large Animal Study. Tissue Eng. Part A 2017, 23, 69–79. [Google Scholar] [CrossRef]
- Wang, N.; Zheng, W.; Cheng, S.; Zhang, W.; Liu, S.; Jiang, X. In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering. Polymers 2017, 9, 318. [Google Scholar] [CrossRef]
- Lee, R.H.; Pulin, A.A.; Seo, M.J.; Kota, D.J.; Ylostalo, J.; Larson, B.L.; Semprun-Prieto, L.; Delafontaine, P.; Prockop, D.J. Intravenous hMSCs Improve Myocardial Infarction in Mice because Cells Embolized in Lung Are Activated to Secrete the Anti-inflammatory Protein TSG-6. Cell Stem Cell 2009, 5, 54–63. [Google Scholar] [CrossRef]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.C.; Geissler, E.K.; Schlitt, H.J.; Baan, C.C.; Dahlke, M.H.; Hoogduijn, M.J. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef]
- Moll, G.; Alm, J.J.; Davies, L.C.; Von Bahr, L.; Heldring, N.; Stenbeck-Funke, L.; Hamad, O.A.; Hinsch, R.; Ignatowicz, L.; Locke, M.; et al. Do Cryopreserved Mesenchymal Stromal Cells Display Impaired Immunomodulatory and Therapeutic Properties? Stem Cells 2014, 32, 2430–2442. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorentz, K.L.; Marini, A.X.; Bruk, L.A.; Gupta, P.; Mandal, B.B.; DiLeo, M.V.; Weinbaum, J.S.; Little, S.R.; Vorp, D.A. Mesenchymal Stem Cell-Conditioned Media-Loaded Microparticles Enhance Acute Patency in Silk-Based Vascular Grafts. Bioengineering 2024, 11, 947. https://doi.org/10.3390/bioengineering11090947
Lorentz KL, Marini AX, Bruk LA, Gupta P, Mandal BB, DiLeo MV, Weinbaum JS, Little SR, Vorp DA. Mesenchymal Stem Cell-Conditioned Media-Loaded Microparticles Enhance Acute Patency in Silk-Based Vascular Grafts. Bioengineering. 2024; 11(9):947. https://doi.org/10.3390/bioengineering11090947
Chicago/Turabian StyleLorentz, Katherine L., Ande X. Marini, Liza A. Bruk, Prerak Gupta, Biman B. Mandal, Morgan V. DiLeo, Justin S. Weinbaum, Steven R. Little, and David A. Vorp. 2024. "Mesenchymal Stem Cell-Conditioned Media-Loaded Microparticles Enhance Acute Patency in Silk-Based Vascular Grafts" Bioengineering 11, no. 9: 947. https://doi.org/10.3390/bioengineering11090947
APA StyleLorentz, K. L., Marini, A. X., Bruk, L. A., Gupta, P., Mandal, B. B., DiLeo, M. V., Weinbaum, J. S., Little, S. R., & Vorp, D. A. (2024). Mesenchymal Stem Cell-Conditioned Media-Loaded Microparticles Enhance Acute Patency in Silk-Based Vascular Grafts. Bioengineering, 11(9), 947. https://doi.org/10.3390/bioengineering11090947