
An Unsupervised Error Detection Methodology for Detecting Mislabels in Healthcare Analytics

 ,
,  , , , , and
, , , , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Dataset and Preprocessing
2.2. Methodology
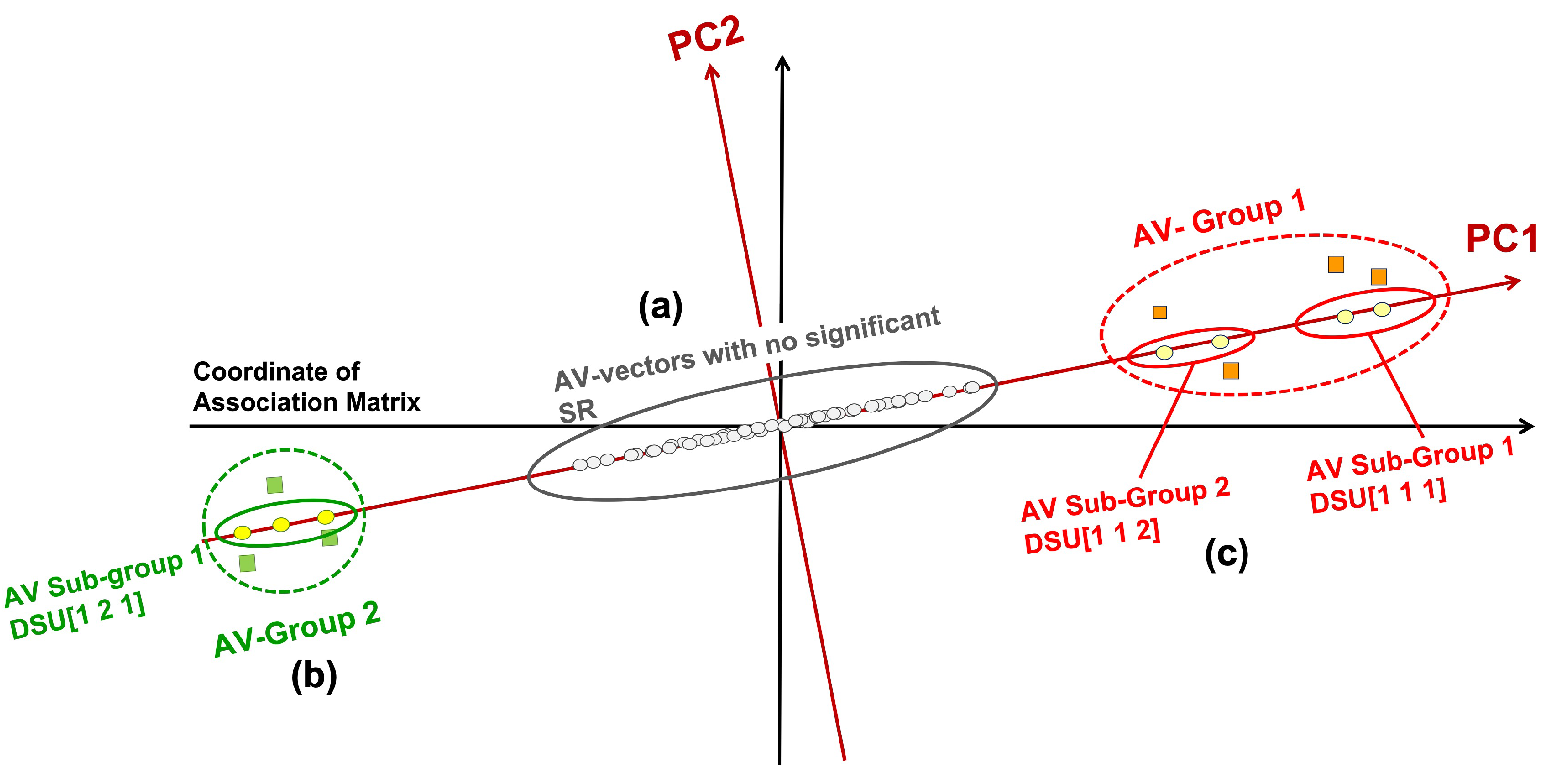
2.2.1. Interpreting Dataset
- Those close to the origin are not statistically significant and thus do not associate with distinct groups/classes (marked as (a) in Figure 1), They are close to the origin because all their coordinates (SRs) are low and insignificant.
- Those at one extremal end of the projections; and those at the opposite extremal end (both marked as (b)).
- The AV groups or subgroups in (b), if their AVs within are statistically connected but disconnected from other groups, may associate with distinct sources (i.e., classes) (marked as (c)).
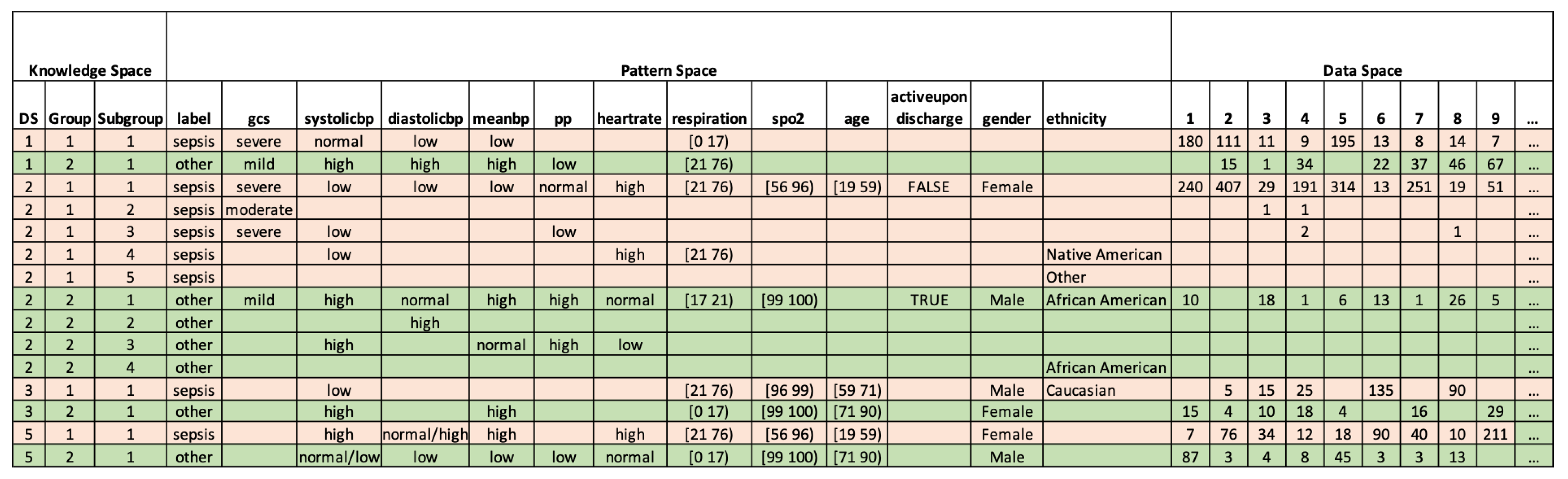
2.2.2. Clustering Patient Records
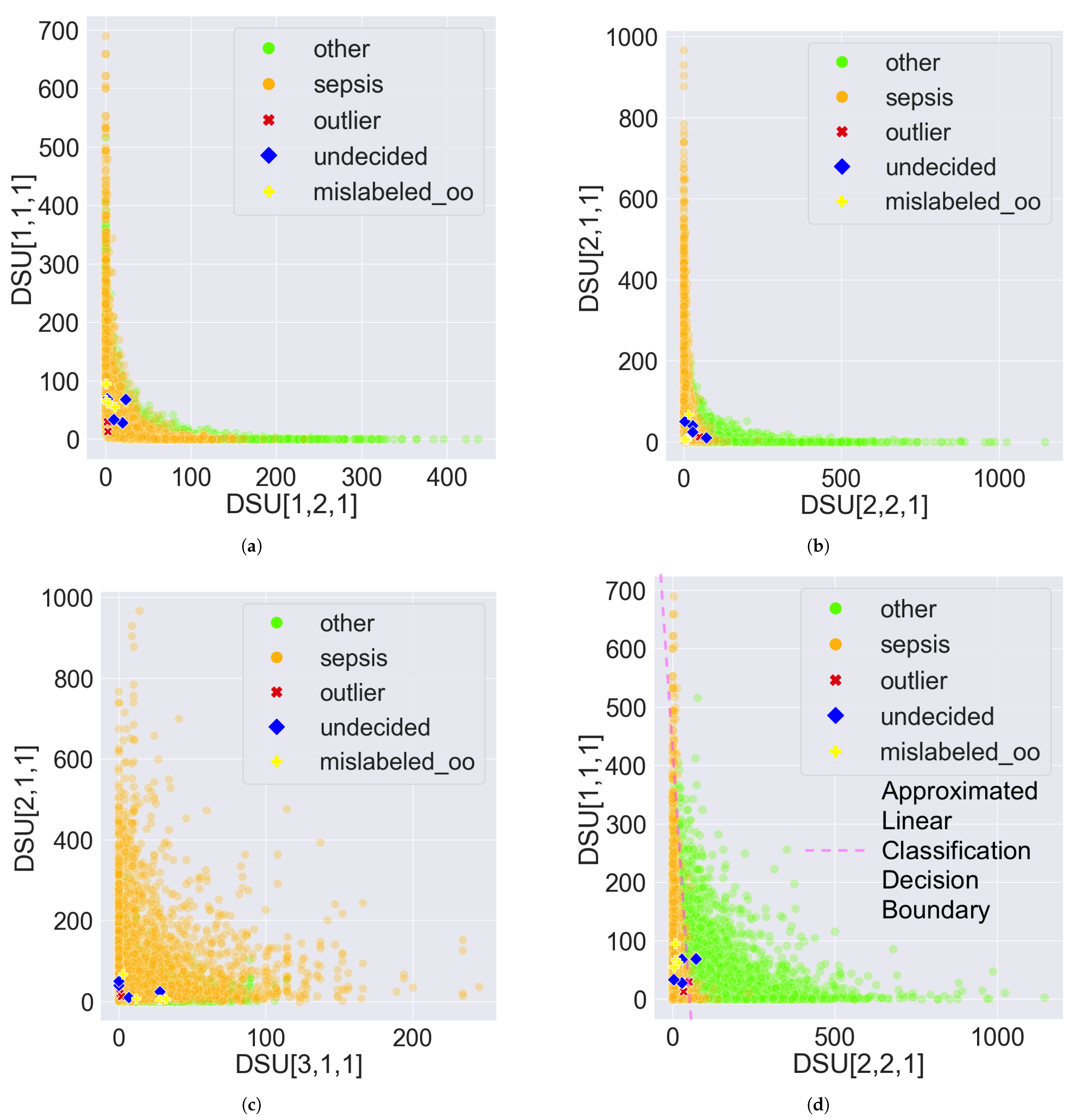
2.2.3. Detecting Abnormal Records
- Mislabelled: If a record is categorized into one class but matches more patterns from a different class according to the PDD output, it suggests the record may be mislabelled. For example, consider the same record with described in Section 2.2.2 with the same setting of the pattern groups where and . If the record is originally labelled as in the dataset, but the relative difference is greater than , this suggests that the record () is more associated with than with . The relative difference is used instead of absolute difference because it provides a scale-independent comparison of the number of patterns associated with one class to another. A value greater than 0.1 indicates that the number of patterns associated with one class is statistically significantly greater than the number associated with another class. Hence, the record () may be mislabelled.
- Outlier: If a record possesses no patterns or very few patterns, it may indicate the record is an outlier. For example, a record with uses the previously described pattern group settings. The comprehensive patterns possessed by this record are: , , , and . Calculating the percentages, is and is . Both percentages are less than or equal to , suggesting that record m possesses fewer than 1% of the patterns associated with either class, which may indicate it is an outlier.
- Undecided: If the number of possessed patterns for a record is similar across different classes, the record should be classified as undecided. For example, a record with uses the previously described pattern group settings. The comprehensive patterns possessed by this record are: , , , and . Calculating the percentages, is the mean of and , which is ; and is the mean of and , which is . Since the difference between the two percentages is zero or less than , record k may be associated with both classes, suggesting it is undecided
3. Results
3.1. Interpretable Results
3.2. Comparison of Unsupervised Learning
3.3. Error Detection Results
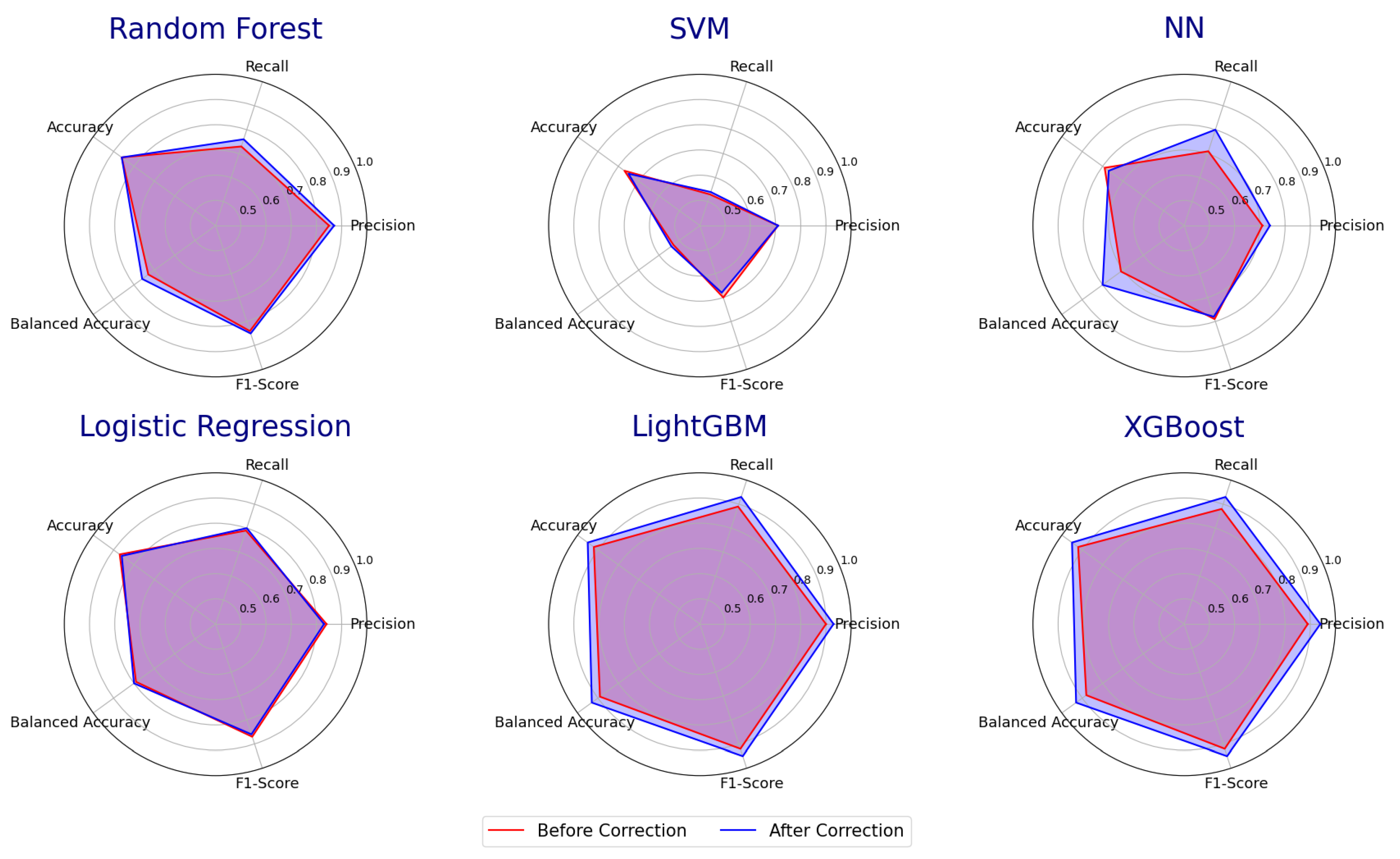
3.4. Error Detection Applied to Supervised Learning Models
4. Discussion
4.1. Interpretability
4.2. Error Detection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ML | Machine Learning |
| EHR | electronic health records |
| PDD | Pattern Discovery and Disentanglement |
| eICU-CRD | eICU Collaborative Research Database |
| GCS | Glasgow coma scale |
| SPO2 | oxygen saturation |
| SR | adjusted standard residual |
| SR-Matrix | statistical residual matrix |
| RSR-Matrix | Reprojected SR-Matrix |
| AV | attribute value |
| PCA | Principal Component Analysis |
| DSU | Disentangled Space Unit |
| SubGroup | Sub-Pattern group |
| Group | Pattern Group |
| PC | Principal Component |
| RF | Random Forests |
Appendix A. Dataset Description and Preprocessing Details
Appendix A.1. Step 1: Generate Temporal Features Based on Vital Signs
Appendix A.2. Step 2: Process Labels
Appendix A.3. Step 2: Filter Features

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Table | Description |
|---|---|---|
| patientUnitStayID | patient | A distinct ICU admission. The same patient can have multiple. |
| observationOffset | vital periodic | The number of minutes, since admission, before the periodic value was recorded. |
| diagnosisoffset | diagnosis | Number of minutes, since admission, before diagnosis was recorded. |
| diagnosisPriority | diagnosis | Diagnosis was marked as Primary, Major, or Other |
| diagnosisString | diagnosis | The full pathstring of diagnosis selected in Philips eCareManager software with sections separated with pipes (e.g., cardiovascular|shock/hypotension|sepsis ) |
| ICD9Code | diagnosis | International Classification of Diseases code used to classify diagnoses (e.g., 518.81, 491.20, etc.) |
Appendix B. Calculation of Adjusted Residual
| Association Table | Systolic—High | Systolic—Low | Total |
|---|---|---|---|
| Diastolic—High | 50 (39) | 40 (51) | 90 |
| Diastolic—Medium | 30 (43.33) | 70 (56.67) | 100 |
| Diastolic—Low | 50 (47.67) | 60 (62.33) | 110 |
| Total | 130 | 170 | 300 |
References
- Weerasinghe, K.; Scahill, S.L.; Pauleen, D.J.; Taskin, N. Big data analytics for clinical decision-making: Understanding health sector perceptions of policy and practice. Technol. Forecast. Soc. Chang. 2022, 174, 121222. [Google Scholar] [CrossRef]
- Rajkomar, A.; Oren, E.; Chen, K.; Dai, A.M.; Hajaj, N.; Hardt, M.; Liu, P.J.; Liu, X.; Marcus, J.; Sun, M.; et al. Scalable and accurate deep learning with electronic health records. NPJ Digit. Med. 2018, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bhimavarapu, U.; Chintalapudi, N.; Battineni, G. Brain tumor detection and categorization with segmentation of improved unsupervised clustering approach and machine learning classifier. Bioengineering 2024, 11, 266. [Google Scholar] [CrossRef]
- Lin, C.Y.; Wu, J.C.H.; Kuan, Y.M.; Liu, Y.C.; Chang, P.Y.; Chen, J.P.; Lu, H.H.S.; Lee, O.K.S. Precision Identification of Locally Advanced Rectal Cancer in Denoised CT Scans Using EfficientNet and Voting System Algorithms. Bioengineering 2024, 11, 399. [Google Scholar] [CrossRef] [PubMed]
- Bedolla, C.N.; Gonzalez, J.M.; Vega, S.J.; Convertino, V.A.; Snider, E.J. An explainable machine-learning model for compensatory reserve measurement: Methods for feature selection and the effects of subject variability. Bioengineering 2023, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Al Sadi, K.; Balachandran, W. Leveraging a 7-Layer Long Short-Term Memory Model for Early Detection and Prevention of Diabetes in Oman: An Innovative Approach. Bioengineering 2024, 11, 379. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.; Chen, C.; Chen, Z.; Huang, H.; Semenova, L.; Zhong, C. Interpretable machine learning: Fundamental principles and 10 grand challenges. Stat. Surv. 2022, 16, 1–85. [Google Scholar] [CrossRef]
- Raghupathi, W.; Raghupathi, V. Big data analytics in healthcare: Promise and potential. Health Inf. Sci. Syst. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Diakiw, S.; VerMilyea, M.; Dinsmore, A.; Perugini, M.; Perugini, D.; Hall, J. Efficient automated error detection in medical data using deep-learning and label-clustering. Sci. Rep. 2023, 13, 19587. [Google Scholar] [CrossRef]
- Caponetto, R.; Fortuna, L.; Graziani, S.; Xibilia, M. Genetic algorithms and applications in system engineering: A survey. Trans. Inst. Meas. Control 1993, 15, 143–156. [Google Scholar] [CrossRef]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef]
- Mehrabi, N.; Morstatter, F.; Saxena, N.; Lerman, K.; Galstyan, A. A survey on bias and fairness in machine learning. ACM Comput. Surv. (CSUR) 2021, 54, 1–35. [Google Scholar] [CrossRef]
- Amann, J.; Blasimme, A.; Vayena, E.; Frey, D.; Madai, V.I.; Consortium, P. Explainability for artificial intelligence in healthcare: A multidisciplinary perspective. BMC Med. Inform. Decis. Mak. 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Sun, Y.; Wong, A.K.; Kamel, M.S. Classification of imbalanced data: A review. Int. J. Pattern Recognit. Artif. Intell. 2009, 23, 687–719. [Google Scholar] [CrossRef]
- Wong, A.K.; Zhou, P.Y.; Butt, Z.A. Pattern discovery and disentanglement on relational datasets. Sci. Rep. 2021, 11, 5688. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Zhou, P.Y.; Lee, A.E.S. Theory and rationale of interpretable all-in-one pattern discovery and disentanglement system. Npj Digit. Med. 2023, 6, 92. [Google Scholar] [CrossRef]
- Ghalwash, M.F.; Radosavljevic, V.; Obradovic, Z. Extraction of interpretable multivariate patterns for early diagnostics. In Proceedings of the 2013 IEEE 13th International Conference on Data Mining, Dallas, TX, USA, 7–10 December 2013; IEEE: Piscataway, NJ, USA, 2013; pp. 201–210. [Google Scholar]
- Metta, C.; Beretta, A.; Pellungrini, R.; Rinzivillo, S.; Giannotti, F. Towards Transparent Healthcare: Advancing Local Explanation Methods in Explainable Artificial Intelligence. Bioengineering 2024, 11, 369. [Google Scholar] [CrossRef]
- Northcutt, C.G.; Athalye, A.; Mueller, J. Pervasive label errors in test sets destabilize machine learning benchmarks. arXiv 2021, arXiv:2103.14749. [Google Scholar]
- Zhou, Z.H. Ensemble Methods: Foundations and Algorithms; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Khan, A.A.; Chaudhari, O.; Chandra, R. A review of ensemble learning and data augmentation models for class imbalanced problems: Combination, implementation and evaluation. Expert Syst. Appl. 2023, 244, 122778. [Google Scholar] [CrossRef]
- Pollard, T.J.; Johnson, A.E.; Raffa, J.D.; Celi, L.A.; Mark, R.G.; Badawi, O. The eICU Collaborative Research Database, a freely available multi-center database for critical care research. Sci. Data 2018, 5, 180178. [Google Scholar] [CrossRef]
- Parmar, S.; Shan, T.; Lee, S.; Kim, Y.; Kim, J.Y. Extending Machine Learning-Based Early Sepsis Detection to Different Demographics. In Proceedings of the 2024 IEEE First International Conference on Artificial Intelligence for Medicine, Health and Care (AIMHC), Laguna Hills, CA, USA, 5–7 February 2024; IEEE: Piscataway, NJ, USA, 2024; pp. 70–71. [Google Scholar]
- Zhou, P.Y.; Sze-To, A.; Wong, A.K. Discovery and disentanglement of aligned residue associations from aligned pattern clusters to reveal subgroup characteristics. BMC Med. Genom. 2018, 11, 35–49. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Hilarius, K.W.; Skippen, P.W.; Kissoon, N. Early recognition and emergency treatment of sepsis and septic shock in children. Pediatr. Emerg. Care 2020, 36, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit. Care Med. 2013, 41, 580–637. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Haixiang, G.; Yijing, L.; Shang, J.; Mingyun, G.; Yuanyue, H.; Bing, G. Learning from class-imbalanced data: Review of methods and applications. Expert Syst. Appl. 2017, 73, 220–239. [Google Scholar] [CrossRef]
- Palczewska, A.; Palczewski, J.; Marchese Robinson, R.; Neagu, D. Interpreting random forest classification models using a feature contribution method. Integr. Reusable Syst. 2014, 263, 193–218. [Google Scholar]
- Marchenko, O.; Russek-Cohen, E.; Levenson, M.; Zink, R.C.; Krukas-Hampel, M.R.; Jiang, Q. Sources of safety data and statistical strategies for design and analysis: Real world insights. Ther. Innov. Regul. Sci. 2018, 52, 170–186. [Google Scholar] [CrossRef]
- Wong, A.K.; Au, W.H.; Chan, K.C. Discovering high-order patterns of gene expression levels. J. Comput. Biol. 2008, 15, 625–637. [Google Scholar] [CrossRef]
- Everitt, B.; Skkrondal, A. The Cambridge Dictionary of Statistics; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Wong, A.K.; Wang, Y. High-order pattern discovery from discrete-valued data. IEEE Trans. Knowl. Data Eng. 1997, 9, 877–893. [Google Scholar] [CrossRef]




| Features | Low Range | Normal Range | High Range |
|---|---|---|---|
| GCS | 3–8 (Severe) | 9–12 (Moderate) | 13–15 (Mild) |
| Systolic BP 1 | <90 mm Hg | 90–120 mm Hg | >120 mm Hg |
| Diastolic BP 1 | <60 mm Hg | 60–80 mm Hg | >80 mm Hg |
| Mean BP 1 | <70 mm Hg | 70–93 mm Hg | >93 mm Hg |
| Pulse Pressure | <30 mm Hg | 30–40 mm Hg | >40 mm Hg |
| Heart Rate | <60 bpm | 60–100 bpm | >100 bpm |
| Unsupervised Learning | Precision | Recall | Accuracy | Balanced Accuracy | Weighted F1-Score |
|---|---|---|---|---|---|
| PDD (28-features data) | 0.83 | 0.90 | 0.87 | 0.89 | 0.88 |
| K-means (28-features data) | 0.43 | 0.42 | 0.52 | 0.42 | 0.54 |
| K-means (114-features data) | 0.59 | 0.51 | 0.75 | 0.51 | 0.67 |
| Classifiers (Before/After Error Detection) | Precision | Recall | Accuracy | Balanced Accuracy | Weighted F1-Score |
|---|---|---|---|---|---|
| Random Forest | 0.85/0.87 | 0.73/0.76 | 0.86/0.86 | 0.73/0.76 | 0.84/0.85 |
| SVM | 0.71/0.71 | 0.53/0.54 | 0.77/0.75 | 0.53/0.54 | 0.7/0.68 |
| NN | 0.71/0.74 | 0.71/0.80 | 0.79/0.77 | 0.71/0.80 | 0.79/0.78 |
| Logistic Regression | 0.84/0.83 | 0.79/0.80 | 0.87/0.86 | 0.79/0.80 | 0.87/0.86 |
| LightGBM | 0.9/0.93 | 0.89/0.93 | 0.92/0.95 | 0.89/0.93 | 0.92/0.95 |
| XGBoost | 0.89/0.94 | 0.88/0.93 | 0.92/0.95 | 0.88/0.93 | 0.92/0.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.-Y.; Lum, F.; Wang, T.J.; Bhatti, A.; Parmar, S.; Dan, C.; Wong, A.K.C. An Unsupervised Error Detection Methodology for Detecting Mislabels in Healthcare Analytics. Bioengineering 2024, 11, 770. https://doi.org/10.3390/bioengineering11080770
Zhou P-Y, Lum F, Wang TJ, Bhatti A, Parmar S, Dan C, Wong AKC. An Unsupervised Error Detection Methodology for Detecting Mislabels in Healthcare Analytics. Bioengineering. 2024; 11(8):770. https://doi.org/10.3390/bioengineering11080770
Chicago/Turabian StyleZhou, Pei-Yuan, Faith Lum, Tony Jiecao Wang, Anubhav Bhatti, Surajsinh Parmar, Chen Dan, and Andrew K. C. Wong. 2024. "An Unsupervised Error Detection Methodology for Detecting Mislabels in Healthcare Analytics" Bioengineering 11, no. 8: 770. https://doi.org/10.3390/bioengineering11080770
APA StyleZhou, P.-Y., Lum, F., Wang, T. J., Bhatti, A., Parmar, S., Dan, C., & Wong, A. K. C. (2024). An Unsupervised Error Detection Methodology for Detecting Mislabels in Healthcare Analytics. Bioengineering, 11(8), 770. https://doi.org/10.3390/bioengineering11080770