
In Vitro Generation of Haploid Germ Cells from Human XY and XXY Immature Testes in a 3D Organoid System

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Testis Material
2.2. Morphologic Testicular Tissue Evaluation
2.3. Cell Isolation, Propagation, Characterization, and Quantification
2.3.1. Isolation and Propagation
2.3.2. Characterization of the Cells in Culture
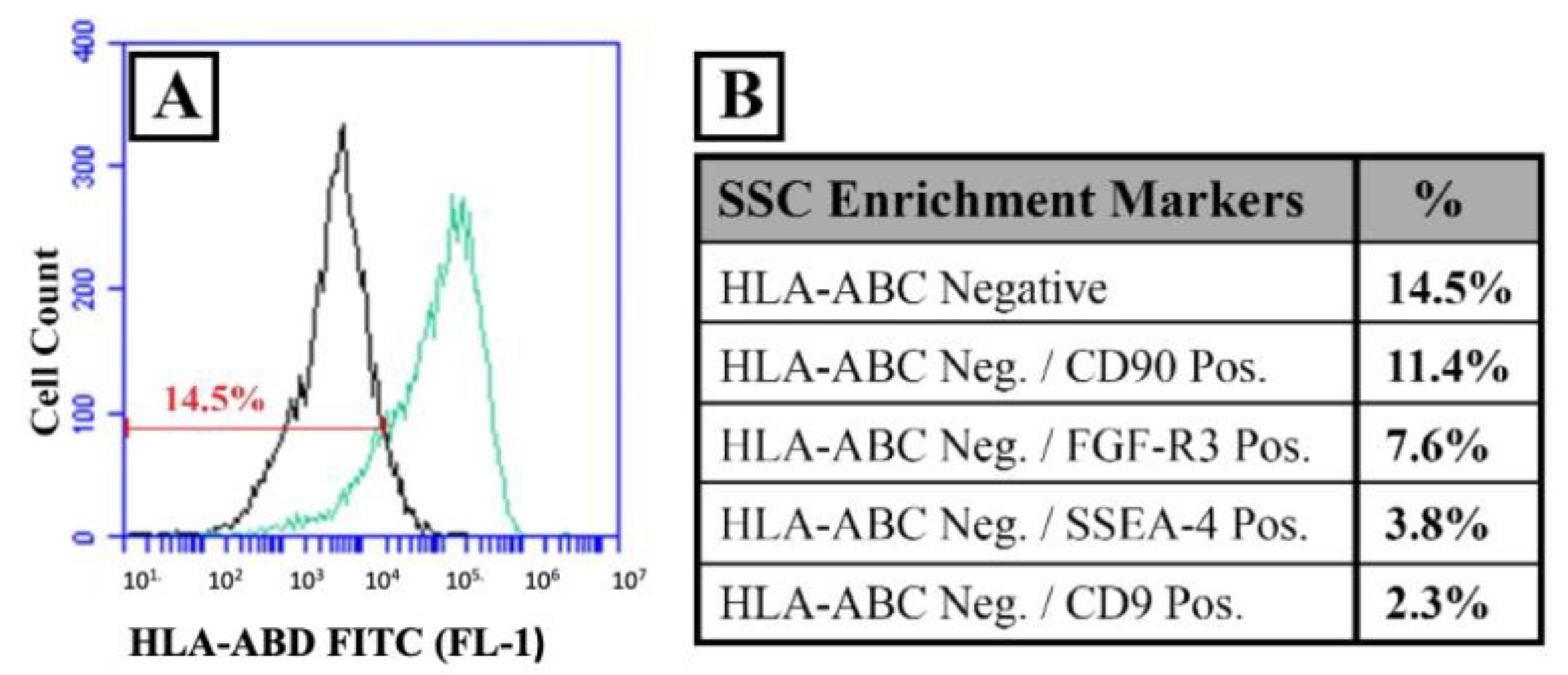
2.3.3. SSC Identification
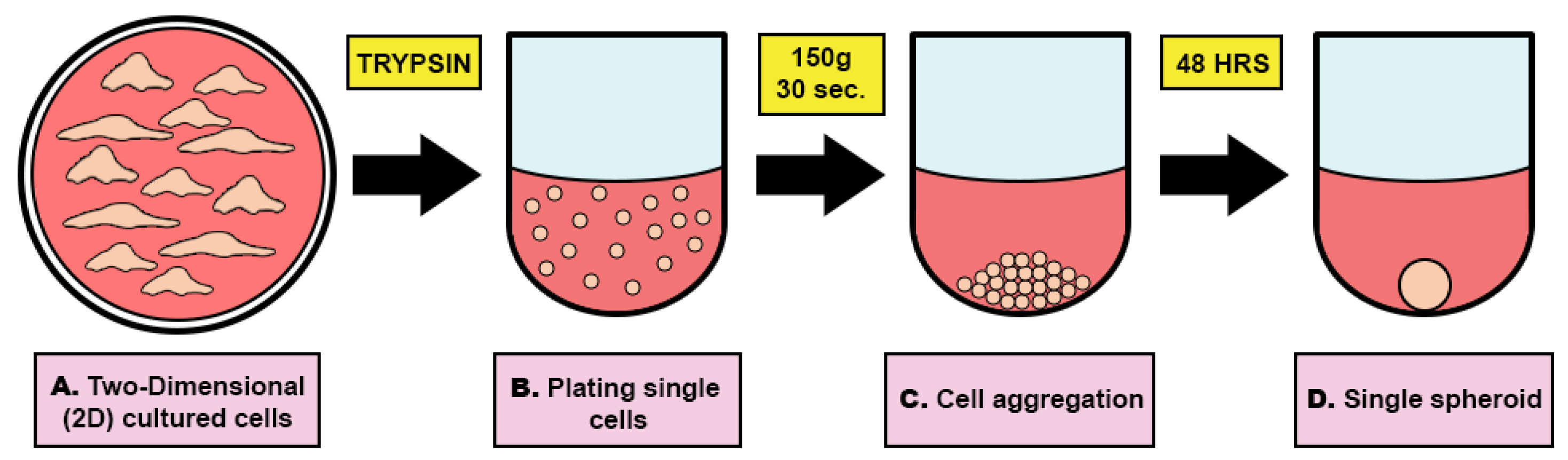
2.4. Human Testicular Organoid (HTO) Formation and Differentiation
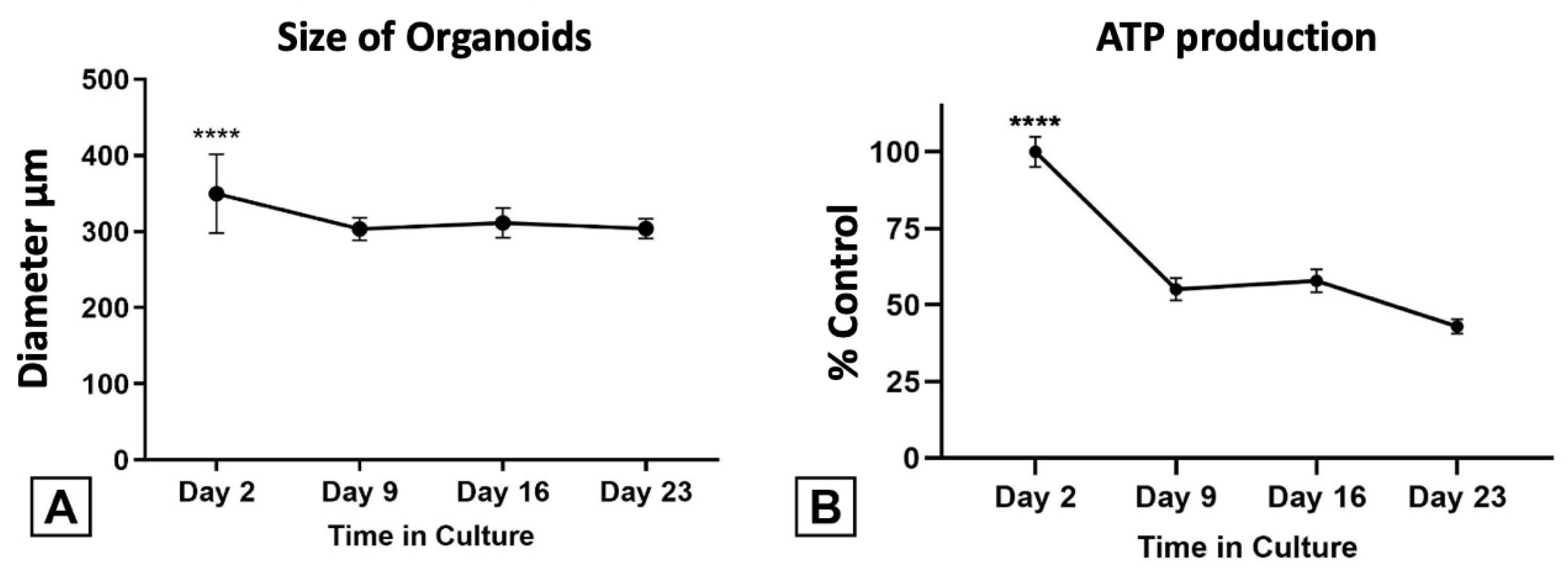
2.4.1. HTO Diameter and Viability
2.4.2. ATP Production
2.4.3. Cell Dissociation and RNA Isolation
2.4.4. Histology Evaluation
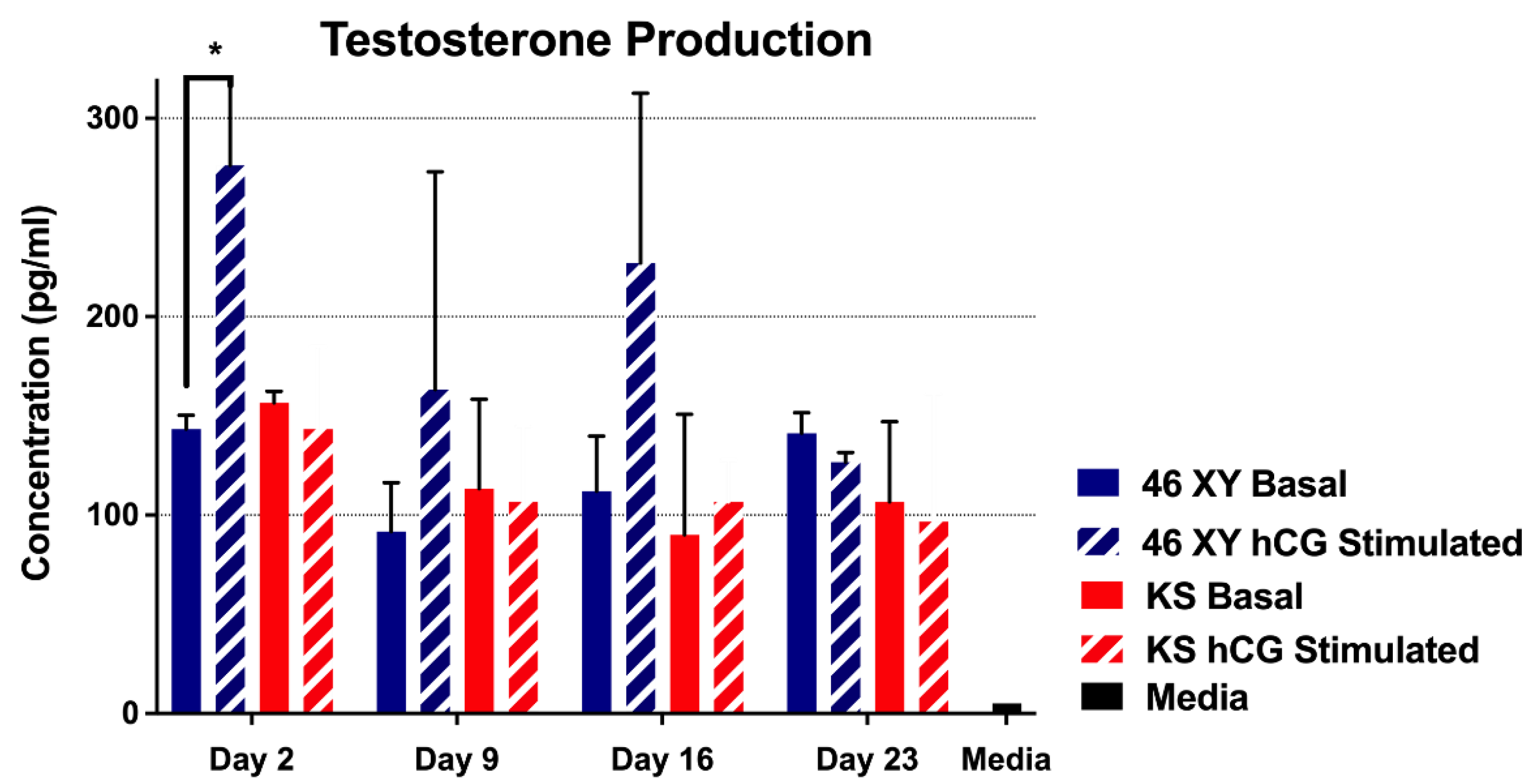
2.4.5. Testosterone Production
2.4.6. Gene Expression Analysis
2.4.7. DNA Fluorescence In Situ Hybridization (FISH) of the HTOs for Ploidy Identification
2.5. Statistics
3. Results
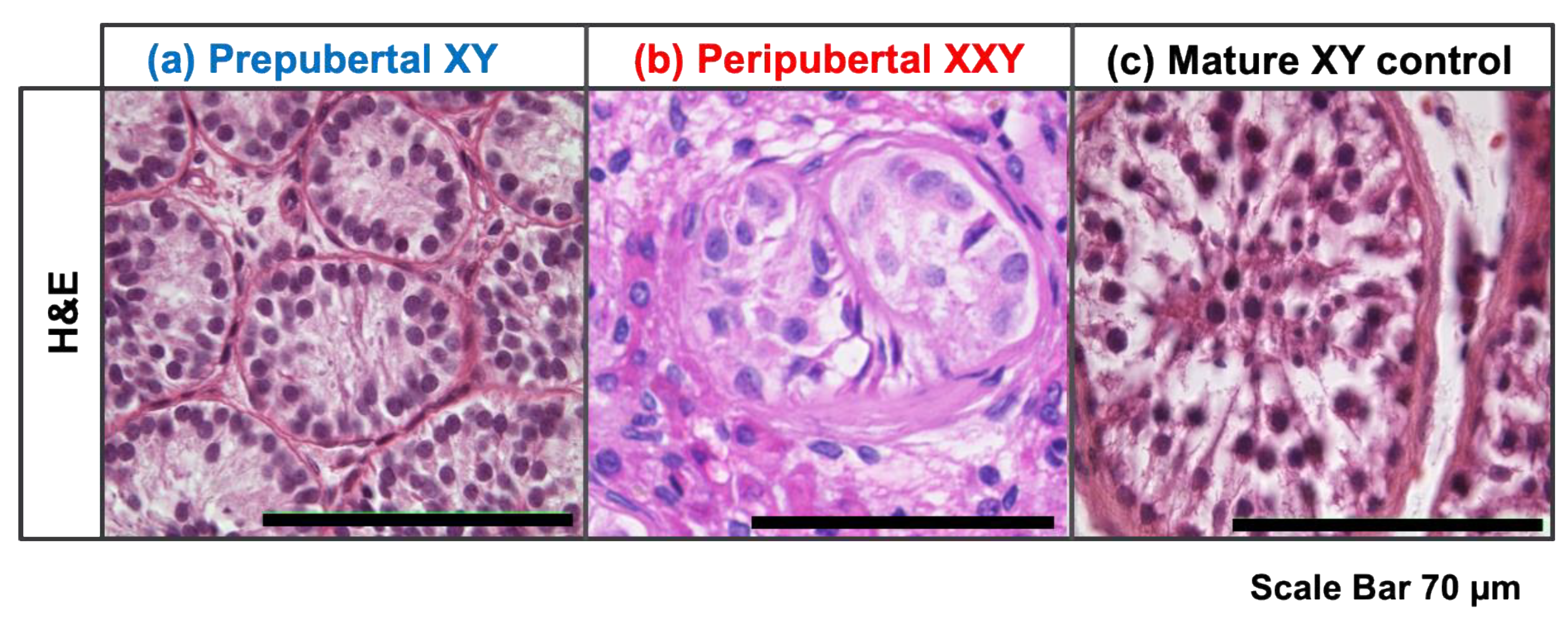
3.1. Immature Testicular Tissue with Neither Differentiating nor Differentiated Germ Cells
3.2. Long-Term Propagation of Testicular Cells
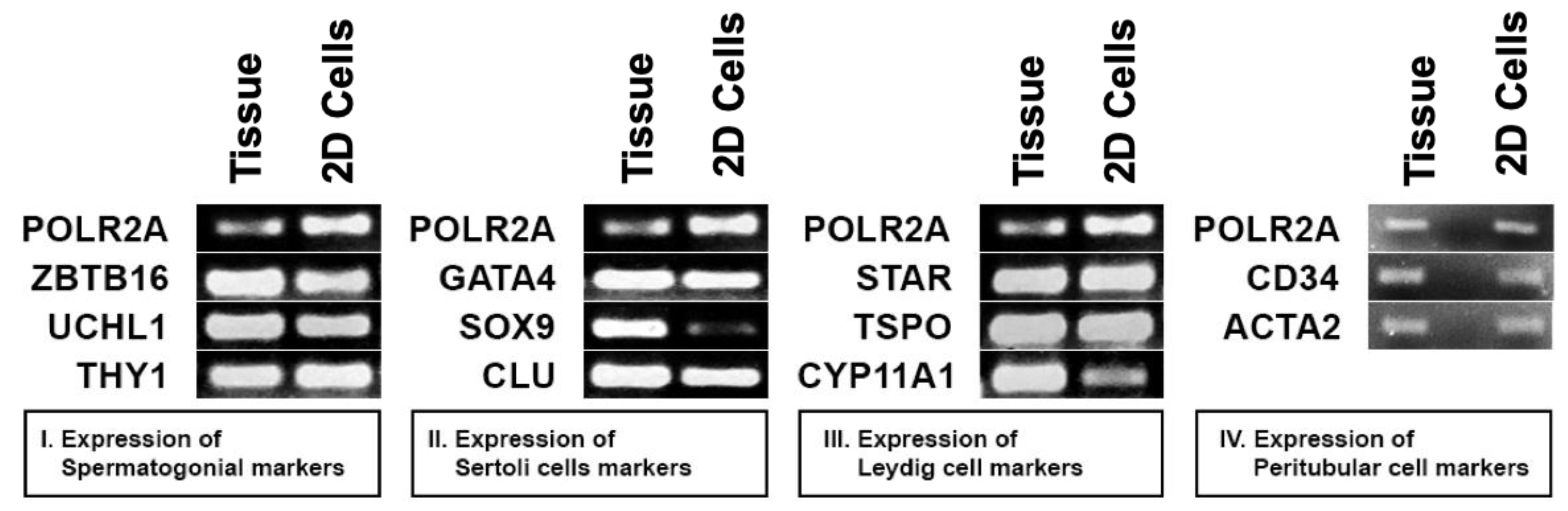
3.3. Presence of All Major Cell Types in Propagated Prepubertal Testicular Cells In Vitro
3.4. HTO Formation and Germ Cell Differentiation
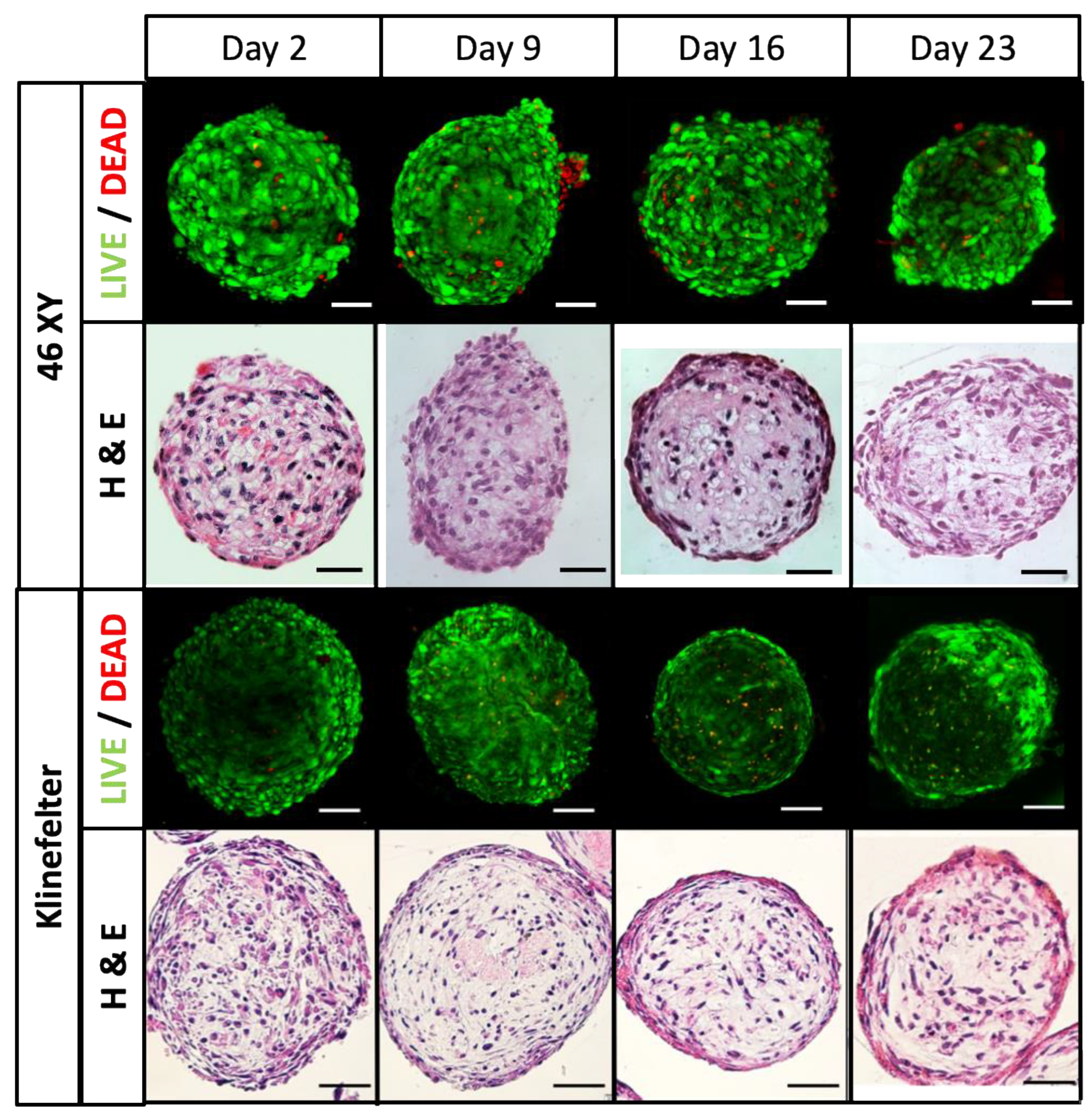
3.4.1. HTOs Maintain Their Structure, Viability, and Function for Three Weeks in Culture
3.4.2. Testosterone Production by HTOs
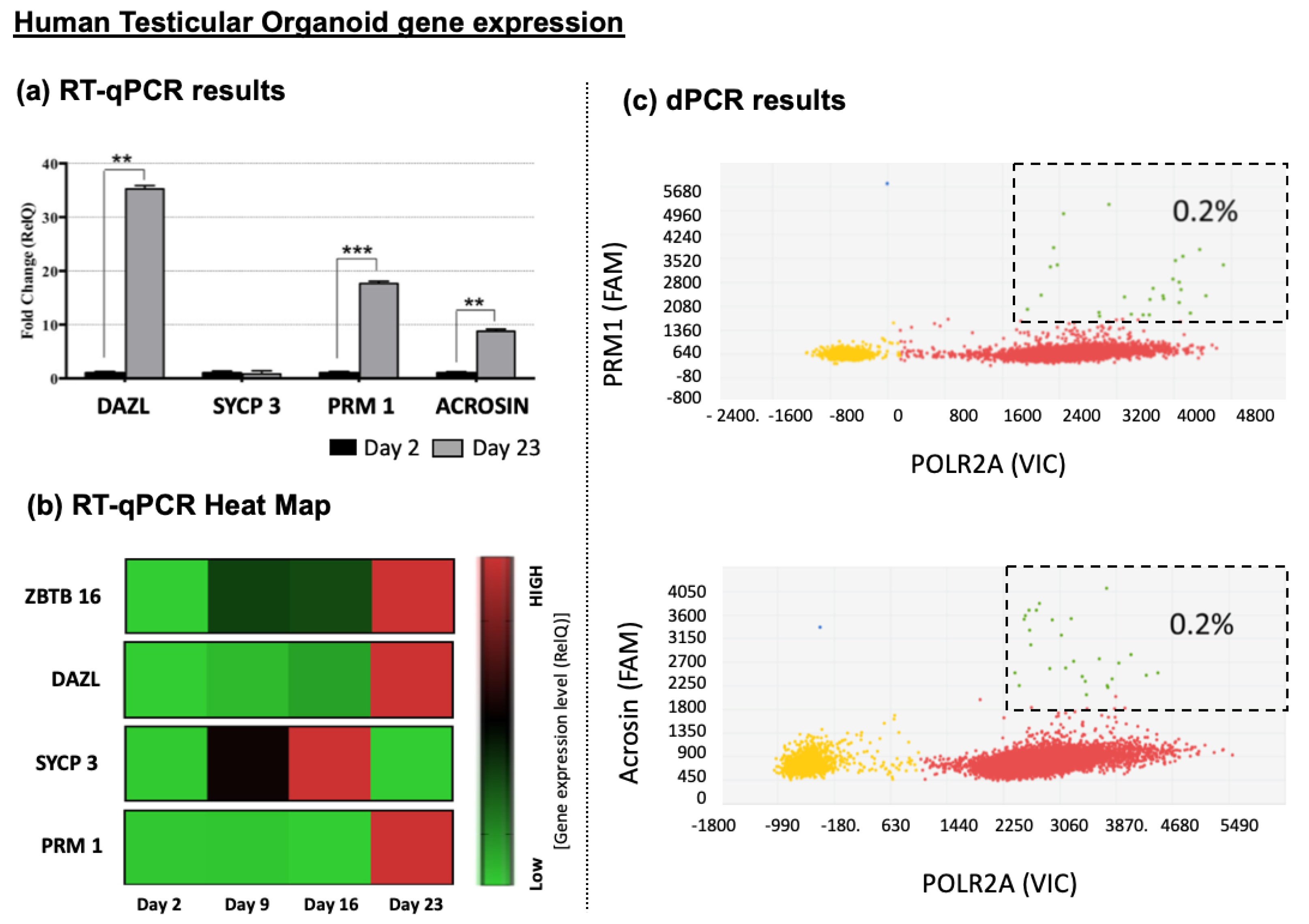
3.4.3. Germ Cells Differentiation of HTOs
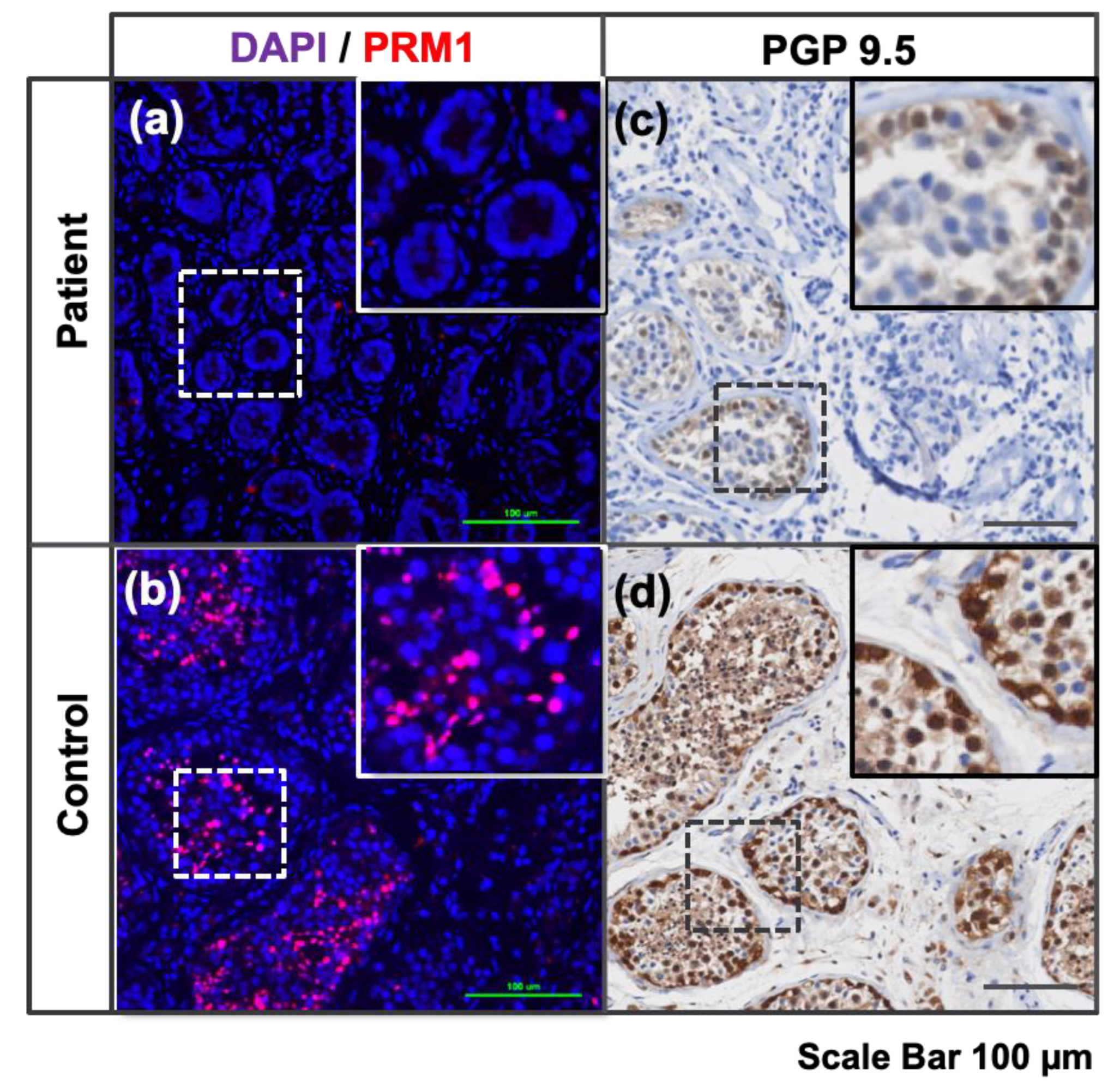
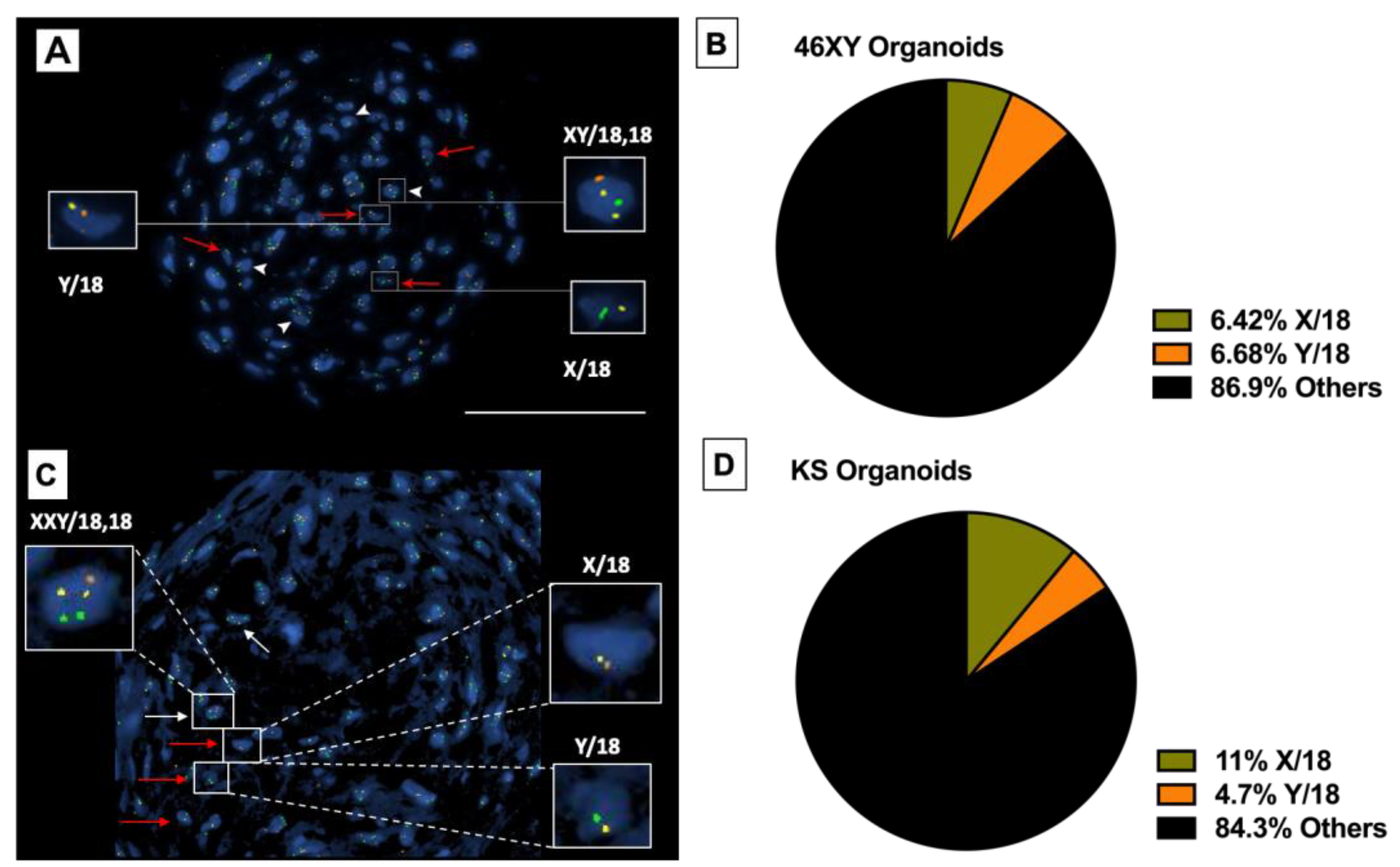
3.4.4. Presence of Haploid Cells inside Differentiated HTOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Felici, M.; Dolci, S. From Testis to Teratomas: A Brief History of Male Germ Cells in Mammals. Int. J. Dev. Biol. 2013, 57, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Eddy, E.M. Regulation of Gene Expression during Spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 451–457. [Google Scholar] [CrossRef]
- Hanukoglu, I. Steroidogenic Enzymes: Structure, Function, and Role in Regulation of Steroid Hormone Biosynthesis. J. Steroid Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Toppari, J. Endocrine, Paracrine and Autocrine Regulation of Testicular Steroidogenesis. Adv. Exp. Med. Biol. 1995, 377, 33–54. [Google Scholar] [CrossRef]
- Oatley, J.M.; Brinster, R.L. The Germline Stem Cell Niche Unit in Mammalian Testes. Physiol. Rev. 2012, 92, 577–595. [Google Scholar] [CrossRef] [PubMed]
- De Kretser, D.M.; Baker, H.W. Infertility in Men: Recent Advances and Continuing Controversies. J. Clin. Endocrinol. Metab. 1999, 84, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, A. Male Subfertility. BMJ 2003, 327, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Baskaran, S.; Parekh, N.; Cho, C.-L.; Henkel, R.; Vij, S.; Arafa, M.; Panner Selvam, M.K.; Shah, R. Male Infertility. Lancet 2021, 397, 319–333. [Google Scholar] [CrossRef]
- Okuyama, N.; Obata, R.; Oka, N.; Nakamura, Y.; Hattori, H.; Nakajo, Y.; Aono, N.; Koizumi, M.; Toya, M.; Nagao, K.; et al. Long-Term Clinical Outcomes of Testicular Sperm Extraction and Intracytoplasmic Sperm Injection for Infertile Men. Reprod. Med. Biol. 2018, 17, 82–88. [Google Scholar] [CrossRef]
- Tournaye, H. Update on Surgical Sperm Recovery—pean View. Hum. Fertil. 2010, 13, 242–246. [Google Scholar] [CrossRef]
- Schrader, M.; Müller, M.; Straub, B.; Miller, K. The Impact of Chemotherapy on Male Fertility: A Survey of the Biologic Basis and Clinical Aspects. Reprod. Toxicol. 2001, 15, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.H.B. Oncofertility and Preservation of Reproductive Capacity in Children and Young Adults. Cancer 2011, 117, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Yumura, Y.; Takeshima, T.; Komeya, M.; Kuroda, S.; Saito, T.; Karibe, J. Fertility and Sexual Dysfunction in Young Male Cancer Survivors. Reprod. Med. Biol. 2022, 21, e12481. [Google Scholar] [CrossRef] [PubMed]
- Lanfranco, F.; Kamischke, A.; Zitzmann, M.; Nieschlag, E. Klinefelter’s Syndrome. Lancet 2004, 364, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Rochira, V.; Pasquali, D.; Balercia, G.; Jannini, E.A.; Ferlin, A.; Klinefelter Italia, N.G. Klinefelter Syndrome (KS): Genetics, Clinical Phenotype and Hypogonadism. J. Endocrinol. Investig. 2017, 40, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bojesen, A.; Juul, S.; Gravholt, C.H. Prenatal and Postnatal Prevalence of Klinefelter Syndrome: A National Registry Study. J. Clin. Endocrinol. Metab. 2003, 88, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gardner, E.J.; Tuke, M.A.; Zhang, H.; Pietzner, M.; Koprulu, M.; Jia, R.Y.; Ruth, K.S.; Wood, A.R.; Beaumont, R.N.; et al. Detection and Characterization of Male Sex Chromosome Abnormalities in the UK Biobank Study. Genet. Med. 2022, 24, 1909–1919. [Google Scholar] [CrossRef]
- Nielsen, J.; Wohlert, M. Sex Chromosome Abnormalities Found among 34,910 Newborn Children: Results from a 13-Year Incidence Study in Arhus, Denmark. Birth Defects Orig. Artic. Ser. 1990, 26, 209–223. [Google Scholar] [PubMed]
- Aksglaede, L.; Wikstrom, A.M.; Rajpert-De Meyts, E.; Dunkel, L.; Skakkebaek, N.E.; Juul, A. Natural History of Seminiferous Tubule Degeneration in Klinefelter Syndrome. Hum. Reprod. Update 2006, 12, 39–48. [Google Scholar] [CrossRef]
- Willems, M.; Gies, I.; Van Saen, D. Germ Cell Loss in Klinefelter Syndrome: When and Why? Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 356–370. [Google Scholar] [CrossRef]
- Corona, G.; Pizzocaro, A.; Lanfranco, F.; Garolla, A.; Pelliccione, F.; Vignozzi, L.; Ferlin, A.; Foresta, C.; Jannini, E.A.; Maggi, M.; et al. Sperm Recovery and ICSI Outcomes in Klinefelter Syndrome: A Systematic Review and Meta-Analysis. Hum. Reprod. Update 2017, 23, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Sermondade, N.; Brioude, F.; Berthaut, I.; Bachelot, A.; Hamid, R.H.; Khattabi, L.E.; Prades, M.; Lévy, R.; Dupont, C. Fertility Preservation in Young Men with Klinefelter Syndrome: A Systematic Review. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 102177. [Google Scholar] [CrossRef] [PubMed]
- Lakhoo, K.; Davies, J.; Chakraborty, S.; Berg, S.; Tennyson, R.; Fowler, D.; Manek, S.; Verrill, C.; Lane, S. Development of a New Reproductive Tissue Cryopreservation Clinical Service for Children: The Oxford Programme. Pediatr. Surg. Int. 2019, 35, 1271–1278. [Google Scholar] [CrossRef]
- Portela, J.M.D.; de Winter-Korver, C.M.; van Daalen, S.K.M.; Meißner, A.; de Melker, A.A.; Repping, S.; van Pelt, A.M.M. Assessment of Fresh and Cryopreserved Testicular Tissues from (Pre)Pubertal Boys during Organ Culture as a Strategy for in Vitro Spermatogenesis. Hum. Reprod. 2019, 34, 2443–2455. [Google Scholar] [CrossRef]
- Tournaye, H.; Dohle, G.R.; Barratt, C.L.R. Fertility Preservation in Men with Cancer. Lancet 2014, 384, 1295–1301. [Google Scholar] [CrossRef]
- Picton, H.M.; Wyns, C.; Anderson, R.A.; Goossens, E.; Jahnukainen, K.; Kliesch, S.; Mitchell, R.T.; Pennings, G.; Rives, N.; Tournaye, H.; et al. A European Perspective on Testicular Tissue Cryopreservation for Fertility Preservation in Prepubertal and Adolescent Boys. Hum. Reprod. 2015, 30, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Valli-Pulaski, H.; Peters, K.A.; Gassei, K.; Steimer, S.R.; Sukhwani, M.; Hermann, B.P.; Dwomor, L.; David, S.; Fayomi, A.P.; Munyoki, S.K.; et al. Testicular Tissue Cryopreservation: 8 Years of Experience from a Coordinated Network of Academic Centers. Hum. Reprod. 2019, 34, 966–977. [Google Scholar] [CrossRef]
- Duffin, K.; Neuhaus, N.; Andersen, C.Y.; Barraud-Lange, V.; Braye, A.; Eguizabal, C.; Feraille, A.; Ginsberg, J.P.; Gook, D.; Goossens, E.; et al. A 20-Year Overview of Fertility Preservation in Boys: New Insights Gained through a Comprehensive International Survey. Hum. Reprod. Open 2024, 2024, hoae010. [Google Scholar] [CrossRef]
- Pendergraft, S.S.; Sadri-Ardekani, H.; Atala, A.; Bishop, C.E. Three-Dimensional Testicular Organoid: A Novel Tool for the Study of Human Spermatogenesis and Gonadotoxicity in Vitro. Biol. Reprod. 2017, 96, 720–732. [Google Scholar] [CrossRef]
- Sun, M.; Yuan, Q.; Niu, M.; Wang, H.; Wen, L.; Yao, C.; Hou, J.; Chen, Z.; Fu, H.; Zhou, F.; et al. Efficient Generation of Functional Haploid Spermatids from Human Germline Stem Cells by Three-Dimensional-Induced System. Cell Death Differ. 2018, 25, 749–766. [Google Scholar] [CrossRef]
- Shetty, G.; Mitchell, J.M.; Meyer, J.M.; Wu, Z.; Lam, T.N.A.; Phan, T.T.; Zhang, J.; Hill, L.; Tailor, R.C.; Peters, K.A.; et al. Restoration of Functional Sperm Production in Irradiated Pubertal Rhesus Monkeys by Spermatogonial Stem Cell Transplantation. Andrology 2020, 8, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Shetty, G.; Mitchell, J.M.; Lam, T.N.A.; Phan, T.T.; Zhang, J.; Tailor, R.C.; Peters, K.A.; Penedo, M.C.; Hanna, C.B.; Clark, A.T.; et al. Postpubertal Spermatogonial Stem Cell Transplantation Restores Functional Sperm Production in Rhesus Monkeys Irradiated before and after Puberty. Andrology 2021, 9, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Fayomi, A.P.; Peters, K.; Sukhwani, M.; Valli-Pulaski, H.; Shetty, G.; Meistrich, M.L.; Houser, L.; Robertson, N.; Roberts, V.; Ramsey, C.; et al. Autologous Grafting of Cryopreserved Prepubertal Rhesus Testis Produces Sperm and Offspring. Science 2019, 363, 1314–1319. [Google Scholar] [CrossRef]
- Ntemou, E.; Kadam, P.; Van Saen, D.; Wistuba, J.; Mitchell, R.T.; Schlatt, S.; Goossens, E. Complete Spermatogenesis in Intratesticular Testis Tissue Xenotransplants from Immature Non-Human Primate. Hum. Reprod. 2019, 34, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Seβenhausen, P.; Gies, I.; Vloeberghs, V.; Tournaye, H.; Van Saen, D.; Goossens, E. Intratesticular Xenografting of Klinefelter Pre-Pubertal Testis Tissue as Potential Model to Study Testicular Fibrosis. Reprod. Biomed. Online 2022, 44, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Katagiri, K.; Kojima, K.; Komeya, M.; Yao, M.; Ogawa, T. In Vitro Spermatogenesis in Explanted Adult Mouse Testis Tissues. PLoS ONE 2015, 10, e0130171. [Google Scholar] [CrossRef]
- Wang, D.; Hildorf, S.; Ntemou, E.; Mamsen, L.S.; Dong, L.; Pors, S.E.; Fedder, J.; Clasen-Linde, E.; Cortes, D.; Thorup, J.; et al. Organotypic Culture of Testicular Tissue from Infant Boys with Cryptorchidism. Int. J. Mol. Sci. 2022, 23, 7975. [Google Scholar] [CrossRef]
- Komeya, M.; Kimura, H.; Nakamura, H.; Yokonishi, T.; Sato, T.; Kojima, K.; Hayashi, K.; Katagiri, K.; Yamanaka, H.; Sanjo, H.; et al. Long-Term Ex Vivo Maintenance of Testis Tissues Producing Fertile Sperm in a Microfluidic Device. Sci. Rep. 2016, 6, 21472. [Google Scholar] [CrossRef]
- Matsumura, T.; Sato, T.; Abe, T.; Sanjo, H.; Katagiri, K.; Kimura, H.; Fujii, T.; Tanaka, H.; Hirabayashi, M.; Ogawa, T. Rat in Vitro Spermatogenesis Promoted by Chemical Supplementations and Oxygen-Tension Control. Sci. Rep. 2021, 11, 3458. [Google Scholar] [CrossRef]
- Perrard, M.-H.; Sereni, N.; Schluth-Bolard, C.; Blondet, A.; D Estaing, S.G.; Plotton, I.; Morel-Journel, N.; Lejeune, H.; David, L.; Durand, P. Complete Human and Rat Ex Vivo Spermatogenesis from Fresh or Frozen Testicular Tissue. Biol. Reprod. 2016, 95, 89. [Google Scholar] [CrossRef]
- de Michele, F.; Poels, J.; Vermeulen, M.; Ambroise, J.; Gruson, D.; Guiot, Y.; Wyns, C. Haploid Germ Cells Generated in Organotypic Culture of Testicular Tissue From Prepubertal Boys. Front. Physiol. 2018, 9, 1413. [Google Scholar] [CrossRef]
- Robinson, M.; Sparanese, S.; Witherspoon, L.; Flannigan, R. Human in Vitro Spermatogenesis as a Regenerative Therapy—Where Do We Stand? Nat. Rev. Urol. 2023, 20, 461–479. [Google Scholar] [CrossRef] [PubMed]
- Komeya, M.; Sato, T.; Ogawa, T. In Vitro Spermatogenesis: A Century-Long Research Journey, Still Half Way Around. Reprod. Med. Biol. 2018, 17, 407–420. [Google Scholar] [CrossRef]
- Easley, C.A.; Phillips, B.T.; McGuire, M.M.; Barringer, J.M.; Valli, H.; Hermann, B.P.; Simerly, C.R.; Rajkovic, A.; Miki, T.; Orwig, K.E.; et al. Direct Differentiation of Human Pluripotent Stem Cells into Haploid Spermatogenic Cells. Cell Rep. 2012, 2, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Ardekani, H.; Mizrak, S.C.; van Daalen, S.K.M.; Korver, C.M.; Roepers-Gajadien, H.L.; Koruji, M.; Hovingh, S.; de Reijke, T.M.; de la Rosette, J.J.M.C.H.; van der Veen, F.; et al. Propagation of Human Spermatogonial Stem Cells in Vitro. JAMA 2009, 302, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Sadri-Ardekani, H.; Akhondi, M.A.; van der Veen, F.; Repping, S.; van Pelt, A.M. In Vitro Propagation of Human Prepubertal Spermatogonial Stem Cells. JAMA 2011, 305, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Galdon, G.; Deebel, N.A.; Zarandi, N.P.; Teramoto, D.; Lue, Y.; Wang, C.; Swerdloff, R.; Pettenati, M.J.; Kearns, W.G.; Howards, S.; et al. In Vitro Propagation of XXY Human Klinefelter Spermatogonial Stem Cells: A Step towards New Fertility Opportunities. Front. Endocrinol. 2022, 13, 1002279. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Orwig, K.E. Spermatogonial Stem Cell Culture in Oncofertility. Urol. Clin. N. Am. 2020, 47, 227–244. [Google Scholar] [CrossRef]
- Kanatsu-Shinohara, M.; Inoue, K.; Ogonuki, N.; Morimoto, H.; Ogura, A.; Shinohara, T. Serum- and Feeder-Free Culture of Mouse Germline Stem Cells. Biol. Reprod. 2011, 84, 97–105. [Google Scholar] [CrossRef]
- Rajan, S.A.P.; Aleman, J.; Wan, M.; Pourhabibi Zarandi, N.; Nzou, G.; Murphy, S.; Bishop, C.E.; Sadri-Ardekani, H.; Shupe, T.; Atala, A.; et al. Probing Prodrug Metabolism and Reciprocal Toxicity with an Integrated and Humanized Multi-Tissue Organ-on-a-Chip Platform. Acta Biomater. 2020, 106, 124–135. [Google Scholar] [CrossRef]
- Skardal, A.; Aleman, J.; Forsythe, S.; Rajan, S.; Murphy, S.; Devarasetty, M.; Pourhabibi Zarandi, N.; Nzou, G.; Wicks, R.; Sadri-Ardekani, H.; et al. Drug Compound Screening in Single and Integrated Multi-Organoid Body-on-a-Chip Systems. Biofabrication 2020, 12, 025017. [Google Scholar] [CrossRef] [PubMed]
- Strange, D.P.; Zarandi, N.P.; Trivedi, G.; Atala, A.; Bishop, C.E.; Sadri-Ardekani, H.; Verma, S. Human Testicular Organoid System as a Novel Tool to Study Zika Virus Pathogenesis. Emerg. Microbes Infect. 2018, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, S.; Strange, D.P.; Jiyarom, B.; Abdelaal, O.; Bradshaw, A.W.; Nerurkar, V.R.; Ward, M.A.; Bakse, J.; Yap, J.; Vanapruks, S.; et al. In Vitro Evidence against Productive SARS-CoV-2 Infection of Human Testicular Cells: Bystander Effects of Infection Mediate Testicular Injury. PLoS Pathog. 2023, 19, e1011409. [Google Scholar] [CrossRef] [PubMed]
- Nickkholgh, B.; Mizrak, S.C.; Korver, C.M.; van Daalen, S.K.; Meissner, A.; Repping, S.; van Pelt, A.M. Enrichment of Spermatogonial Stem Cells from Long-Term Cultured Human Testicular Cells. Fertil. Steril. 2014, 102, 558–565.e5. [Google Scholar] [CrossRef] [PubMed]
- Nickkholgh, B.; Mizrak, S.C.; van Daalen, S.K.M.; Korver, C.M.; Sadri-Ardekani, H.; Repping, S.; van Pelt, A.M.M. Genetic and Epigenetic Stability of Human Spermatogonial Stem Cells during Long-Term Culture. Fertil. Steril. 2014, 102, 1700–1707.e1. [Google Scholar] [CrossRef] [PubMed]
- Zohni, K.; Zhang, X.; Tan, S.L.; Chan, P.; Nagano, M. CD9 Is Expressed on Human Male Germ Cells That Have a Long-Term Repopulation Potential after Transplantation into Mouse Testes. Biol. Reprod. 2012, 87, 27. [Google Scholar] [CrossRef] [PubMed]
- Hutter, H.; Dohr, G. HLA Expression on Immature and Mature Human Germ Cells. J. Reprod. Immunol. 1998, 38, 101–122. [Google Scholar] [CrossRef]
- Griswold, M.D.; Oatley, J.M. Concise Review: Defining Characteristics of Mammalian Spermatogenic Stem Cells. Stem. Cells 2013, 31, 8–11. [Google Scholar] [CrossRef]
- Guo, Y.; Hai, Y.; Gong, Y.; Li, Z.; He, Z. Characterization, Isolation, and Culture of Mouse and Human Spermatogonial Stem Cells. J. Cell Physiol. 2014, 229, 407–413. [Google Scholar] [CrossRef]
- Baert, Y.; Stukenborg, J.-B.; Landreh, M.; De Kock, J.; Jörnvall, H.; Söder, O.; Goossens, E. Derivation and Characterization of a Cytocompatible Scaffold from Human Testis. Hum. Reprod. 2015, 30, 256–267. [Google Scholar] [CrossRef]
- Sato, T.; Katagiri, K.; Gohbara, A.; Inoue, K.; Ogonuki, N.; Ogura, A.; Kubota, Y.; Ogawa, T. In Vitro Production of Functional Sperm in Cultured Neonatal Mouse Testes. Nature 2011, 471, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Yokonishi, T.; Sato, T.; Katagiri, K.; Ogawa, T. In Vitro Spermatogenesis Using an Organ Culture Technique. Methods Mol. Biol. 2013, 927, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Hogg, K.; Western, P.S. Differentiation of Fetal Male Germline and Gonadal Progenitor Cells Is Disrupted in Organ Cultures Containing Knockout Serum Replacement. Stem. Cells Dev. 2015, 24, 2899–2911. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Manufacturer | Catalogue # | Final Conc. | StemPro Complete | Formation Media | Differentiating Media |
|---|---|---|---|---|---|---|
| Bovine Serum Albumin | Sigma-Aldrich | A3059 | 5 mg/mL | ✓ | ✓ | ✓ |
| D (+) Glucose | Sigma-Aldrich | G7021 | 6 mg/mL | ✓ | ✓ | ✓ |
| Ascorbic acid | Sigma-Aldrich | A4544 | 1 × 10−4 M | ✓ | ✓ | ✓ |
| Transferrin (Apo) | Sigma-Aldrich | T1147 | 100 µg/mL | ✓ | ✓ | ✓ |
| Pyruvic Acid | Sigma-Aldrich | P2256 | 30 mg/mL | ✓ | ✓ | ✓ |
| d-Biotin | Sigma-Aldrich | B4501 | 10 µg/mL | ✓ | ✓ | ✓ |
| 2β-mercaptoethanol | Sigma-Aldrich | M3148 | 5 × 10−5 M | ✓ | ✓ | ✓ |
| DL-Lactic Acid | Sigma-Aldrich | L4263 | 1 µL/mL | ✓ | ✓ | ✓ |
| MEM NEAA | Invitrogen | 11140-050 | 10 µL/mL | ✓ | ✓ | ✓ |
| Stem Pro-34 Supplement | Gibco, Thermofisher | 10641-025 | 26 µL/mL | ✓ | ✓ | ✓ |
| Insulin | Cell Applications | 128-100 | 25 µg/mL | ✓ | ✓ | ✓ |
| Sodium Selenite | Sigma-Aldrich | S1382 | 30 nM | ✓ | ✓ | ✓ |
| Putrescine | Sigma-Aldrich | P7505 | 60 µM | ✓ | ✓ | ✓ |
| L-Glutamin | Invitrogen | 25030-081 | 2 mM | ✓ | ✓ | ✓ |
| MEM Vitamin Solution | Invitrogen | 11120-052 | 10 µL/mL | ✓ | ✓ | ✓ |
| Β-Estradiol | Sigma-Aldrich | E2758 | 30 ng/mL | ✓ | ✓ | ✓ |
| Progesterone | Sigma-Aldrich | P8783 | 60 ng/mL | ✓ | ✓ | ✓ |
| Epidermal Growth Factor (EGF) | Sigma-Aldrich | E9644 | 20 ng/mL | ✓ | ✓ | ✓ |
| Human basic Fibroblast Growth Factor (hbFGF) | Sigma-Aldrich | F0291 | 10 ng/mL | ✓ | ✓ | ✓ |
| Glial Cell Line-Derived Neurotrophic Factor (GDNF) | Sigma-Aldrich | G1777 | 10 ng/mL | ✓ | ✓ | ✓ |
| Leukemia Inhibitor Factor | Sigma-Aldrich | LIF1010 | 10 ng/mL | ✓ | ✓ | ✕ |
| Fetal Calf Serum (FCS) | Invitrogen | 10437010 | 1% | ✓ | 30% | ✓ |
| Penicilline/Streptomycine (Pen/Strep) | Invitrogen | 15140-122 | 0.50% | ✓ | ✓ | ✓ |
| Gentamycin | Gibco, Thermofisher | 15750078 | 50 µg/mL | ✕ | ✓ | ✓ |
| Recombinant human stem cell factor (SCF) | Peprotech | 300-07 | 100 ng/mL | ✕ | ✕ | ✓ |
| Retinoic Acid | Sigma-Aldrich | R2625 | 10 µM | ✕ | ✕ | ✓ |
| Human chorionic gonadotropin (hCG) | Sigma-Aldrich | C8554 | 1 mIU/mL | ✕ | ✕ | ✓ |
| Follicle Stimulating Hormone (FSH) | Sigma-Aldrich | F4021 | 2.5 × 10−5 IU/mL | ✕ | ✕ | ✓ |
| Human testis Extra-Cellular Matrix (ECM) | n/a | n/a | 1 µg/mL | ✕ | ✓ | ✓ |
| Gene ID | Amplicon Length (Base Pair) | Catalog # |
|---|---|---|
| ACROSIN | 144 | 4331182 (Hs00356147_m1) |
| PRM1 | 99 | 4331182 (Hs00358158_g1) |
| ZBTB 16 (PLZF) | 65 | 4331182 (Hs00957433_m1) |
| UCHL 1 (PGP9.5) | 80 | 4331182 (Hs00985157_m1) |
| THY1 (CD-90) | 60 | 4331182 (Hs009174816_m1) |
| GATA 4 | 68 | 4331182 (Hs00171403_m1) |
| CLUSTERIN | 93 | 4331182 (Hs00971656_m1) |
| SOX 9 | 101 | 4331182 (Hs01001343_g1) |
| STAR | 85 | 4331182 (Hs00264912_m1) |
| TSPO | 57 | 4331182 (Hs00559362_m1) |
| CYP11A1 | 81 | 4331182 (Hs00897320_m1) |
| CD34 | 63 | 4448892 (Hs02576480_m1) |
| ACTA2 | 64 | 4331182 (Hs00909449_m1) |
| POLR2A | 61 | 4331182 (Hs00172187_m1)4448484 (Hs00172187_m1) |
| Specificity | Host | Type | Fluorochrome | Manufacturer | Catalog # |
|---|---|---|---|---|---|
| HLA-ABC | Mouse | Monoclonal Anti-Human | APC | BD Pharmigen | 555555 |
| HLA-ABC | Mouse | Monoclonal Anti-Human | FITC | BD Pharmigen | 555552 |
| SSEA-4 | Mouse | Monoclonal Anti-Human/Mouse | FITC | R&D Systems | FAB1435F |
| FGF-R3 | Mouse | Monoclonal Anti-Human | PE | R&D Systems | FAB766P |
| CD-9 | Mouse | Monoclonal Anti-Human | PerCP-Cy5.5 | BD Pharmigen | 341649 |
| CD-90 | Mouse | Monoclonal Anti-Human | PE | BD Pharmigen | 555596 |
| Control | Mouse | IgG1 Isotype | FITC | BD Pharmigen | 340755 |
| Control | Mouse | IgG1 Isotype | PerCP-Cy5.5 | BD Pharmigen | 347212 |
| Control | Mouse | IgG1 Isotype | APC | BD Pharmigen | 340754 |
| Control | Mouse | IgG1 Isotype | PE | BD Pharmigen | 340761 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galdon, G.; Zarandi, N.P.; Deebel, N.A.; Zhang, S.; Cornett, O.; Lyalin, D.; Pettenati, M.J.; Lue, Y.; Wang, C.; Swerdloff, R.; et al. In Vitro Generation of Haploid Germ Cells from Human XY and XXY Immature Testes in a 3D Organoid System. Bioengineering 2024, 11, 677. https://doi.org/10.3390/bioengineering11070677
Galdon G, Zarandi NP, Deebel NA, Zhang S, Cornett O, Lyalin D, Pettenati MJ, Lue Y, Wang C, Swerdloff R, et al. In Vitro Generation of Haploid Germ Cells from Human XY and XXY Immature Testes in a 3D Organoid System. Bioengineering. 2024; 11(7):677. https://doi.org/10.3390/bioengineering11070677
Chicago/Turabian StyleGaldon, Guillermo, Nima Pourhabibi Zarandi, Nicholas A. Deebel, Sue Zhang, Olivia Cornett, Dmitry Lyalin, Mark J. Pettenati, YanHe Lue, Christina Wang, Ronald Swerdloff, and et al. 2024. "In Vitro Generation of Haploid Germ Cells from Human XY and XXY Immature Testes in a 3D Organoid System" Bioengineering 11, no. 7: 677. https://doi.org/10.3390/bioengineering11070677
APA StyleGaldon, G., Zarandi, N. P., Deebel, N. A., Zhang, S., Cornett, O., Lyalin, D., Pettenati, M. J., Lue, Y., Wang, C., Swerdloff, R., Shupe, T. D., Bishop, C., Stogner, K., Kogan, S. J., Howards, S., Atala, A., & Sadri-Ardekani, H. (2024). In Vitro Generation of Haploid Germ Cells from Human XY and XXY Immature Testes in a 3D Organoid System. Bioengineering, 11(7), 677. https://doi.org/10.3390/bioengineering11070677