
Enhancement of Classifier Performance with Adam and RanAdam Hyper-Parameter Tuning for Lung Cancer Detection from Microarray Data—In Pursuit of Precision


Abstract
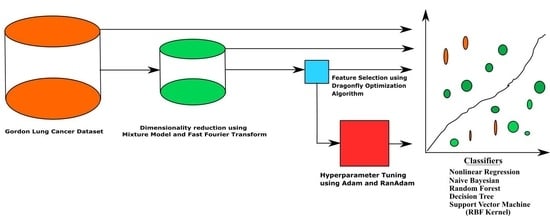
1. Introduction
Review of Previous Work
2. Materials and Methods
2.1. Details about the Dataset
2.2. Dimensionality Reduction (DimRe)
2.2.1. Mixture Model for DimRe
2.2.2. Fast Fourier Transform for DimRe
2.2.3. Impact Analysis of DimRe Methods through Statistics
2.3. Feature Selection (FS) Techniques
2.4. Classification
2.4.1. Nonlinear Regression
2.4.2. Naive Bayesian Classifier
2.4.3. Decision Tree Classifier
2.4.4. Random Forest
2.4.5. SVM (RBF)
2.5. Training and Testing
3. Results and Discussion
3.1. Hyper-Parameter Tuning
3.1.1. Adam Hyper-Parameter Tuning
| Algorithm 1. Adam Hyper-parameter Tuning |
|
3.1.2. RanAdam Hyper-Parameter Tuning
| Algorithm 2. RanAdam Hyper-parameter Tuning |
|
3.2. Computational Complexity (CC)
4. Limitations
5. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644. [Google Scholar] [CrossRef] [PubMed]
- Schabath, M.B.; Cote, M.L. Cancer progress and priorities: Lung cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta BBA-Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Azizi, A.J.; IIIzam, E.; Nazirah, A.; Sharifa, S.; Abbas, S. Lung cancer: Risk factors, management, and prognosis. IOSR J. Dent. Med. Sci. 2016, 15, 94–101. [Google Scholar] [CrossRef]
- Causey, J.L.; Zhang, J.; Ma, S.; Jiang, B.; Qualls, J.A.; Politte, D.G.; Prior, F.; Zhang, S.; Huang, X. Highly accurate model for prediction of lung nodule malignancy with CT scans. Sci. Rep. 2018, 8, 9286. [Google Scholar] [CrossRef] [PubMed]
- Mukae, H.; Kaneko, T.; Obase, Y.; Shinkai, M.; Katsunuma, T.; Takeyama, K.; Terada, J.; Niimi, A.; Matsuse, H.; Yatera, K.; et al. The Japanese respiratory society guidelines for the management of cough and sputum (digest edition). Respir. Investig. 2021, 59, 270–290. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef]
- Leong, S.; Shaipanich, T.; Lam, S.; Yasufuku, K. Diagnostic bronchoscopy—Current and future perspectives. J. Thorac. Dis. 2013, 5, S498–S510. [Google Scholar] [CrossRef]
- Visser, E.P.; Disselhorst, J.A.; Brom, M.; Laverman, P.; Gotthardt, M.; Oyen, W.J.; Boerman, O.C. Spatial resolution and sensitivity of the Inveon small-animal PET scanner. J. Nucl. Med. 2009, 50, 139–147. [Google Scholar] [CrossRef]
- Rivera, M.P.; Mehta, A.C.; Wahidi, M.M. Establishing the diagnosis of lung cancer: Diagnosis and management of lung cancer: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013, 143, e142S–e165S. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, C.C.; Ugras, S.K.; Kazam, J.J.; Zhu, B.; Scognamiglio, T.; Chen, Y.-T.; Fahey, T.J. Microarray analysis of thyroid nodule fine-needle aspirates accurately classifies benign and malignant lesions. J. Mol. Diagn. 2006, 8, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Dhaun, N.; Bellamy, C.O.; Cattran, D.C.; Kluth, D.C. Utility of renal biopsy in the clinical management of renal disease. Kidney Int. 2014, 85, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.V.; Rocke, D.M. Tumor classification by partial least squares using microarray gene expression data. Bioinformatics 2002, 18, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Saheed, Y.K. Effective dimensionality reduction model with machine learning classification for microarray gene expression data. In Data Science for Genomics; Academic Press: Cambridge, MA, USA, 2023; pp. 153–164. [Google Scholar]
- Jaeger, J.; Sengupta, R.; Ruzzo, W.L. Improved gene selection for classification of microarrays. Biocomputing 2002, 2003, 53–64. [Google Scholar]
- De Souza, J.T.; De Francisco, A.C.; De Macedo, D.C. Dimensionality reduction in gene expression data sets. IEEE Access 2019, 7, 61136–61144. [Google Scholar] [CrossRef]
- Rafique, O.; Mir, A. Weighted dimensionality reduction and robust Gaussian mixture model based cancer patient subtyping from gene expression data. J. Biomed. Inform. 2020, 112, 103620. [Google Scholar] [CrossRef]
- Inamura, K.; Fujiwara, T.; Hoshida, Y.; Isagawa, T.; Jones, M.H.; Virtanen, C.; Shimane, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; et al. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrix factorization. Oncogene 2005, 24, 7105–7113. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Huang, P.Y.; Chen, D.T. Sparse principal component analysis in cancer research. Transl. Cancer Res. 2014, 3, 182. [Google Scholar]
- Mollaee, M.; Moattar, M.H. A novel feature extraction approach based on ensemble feature selection and modified discriminant independent component analysis for microarray data classification. Biocybern. Biomed. Eng. 2016, 36, 521–529. [Google Scholar] [CrossRef]
- Chen, J.W.; Dhahbi, J. Lung adenocarcinoma and lung squamous cell carcinoma cancer classification, biomarker identification, and gene expression analysis using overlapping feature selection methods. Sci. Rep. 2021, 11, 13323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Y.; Takagi, T.; Song, J.; Tian, Y.-S.; Shibuya, T. Genetic algorithm-based feature selection with manifold learning for cancer classification using microarray data. BMC Bioinform. 2023, 24, 139. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Rodriguez, C.; Madabhushi, A. Investigating the efficacy of nonlinear dimensionality reduction schemes in classifying gene and protein expression studies. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008, 5, 368–384. [Google Scholar] [CrossRef] [PubMed]
- Raweh, A.A.; Nassef, M.; Badr, A. A Hybridized Feature Selection and Extraction Approach for Enhancing Cancer Prediction Based on DNA Methylation. IEEE Access 2018, 6, 15212–15223. [Google Scholar] [CrossRef]
- Otoom, A.F.; Abdallah, E.E.; Hammad, M. Breast cancer classification: Comparative performance analysis of image shape-based features and microarray gene expression data. Int. J. Bio-Sci. Bio-Technol. 2015, 7, 37–46. [Google Scholar] [CrossRef]
- Orsenigo, C.; Vercellis, C. A comparative study of nonlinear manifold learning methods for cancer microarray data classification. Expert Syst. Appl. 2013, 40, 2189–2197. [Google Scholar] [CrossRef]
- Fan, L.; Poh, K.-L.; Zhou, P. A sequential feature extraction approach for naïve bayes classification of microarray data. Expert Syst. Appl. 2009, 36, 9919–9923. [Google Scholar] [CrossRef]
- Chen, K.-H.; Wang, K.-J.; Angelia, M.-A. Applying particle swarm optimization-based decision tree classifier for cancer classification on gene expression data. Appl. Soft Comput. 2014, 24, 773–780. [Google Scholar] [CrossRef]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef]
- Azzawi, H.; Hou, J.; Xiang, Y.; Alanni, R. Lung cancer prediction from microarray data by gene expression programming. IET Syst. Biol. 2016, 10, 168–178. [Google Scholar] [CrossRef]
- Kotsiantis, S.B.; Zaharakis, I.D.; Pintelas, P.E. Machine learning: A review of classification and combining techniques. Artif. Intell. Rev. 2006, 26, 159–190. [Google Scholar] [CrossRef]
- Ioannou, G.; Tagaris, T.; Stafylopatis, A. AdaLip: An Adaptive Learning Rate Method per Layer for Stochastic Optimization. Neural Process. Lett. 2023, 55, 6311–6338. [Google Scholar] [CrossRef]
- Alrefai, N.; Ibrahim, O. Optimized feature selection method using particle swarm intelligence with ensemble learning for cancer classification based on microarray datasets. Neural Comput. Appl. 2022, 34, 13513–13528. [Google Scholar] [CrossRef]
- Quitadadmo, A.; Johnson, J.; Shi, X. Bayesian hyperparameter optimization for machine learning based eQTL analysis. In Proceedings of the BCB 17: 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017; pp. 98–106. [Google Scholar]
- Wisesty, U.N.; Sthevanie, F.; Rismala, R. Momentum Backpropagation Optimization for Cancer Detection Based on DNA Microarray Data. Int. J. Artif. Intell. Res. 2020, 4, 127–134. [Google Scholar] [CrossRef]
- Rakshitha, K.P.; Naveen, N.C. Op-RMSprop (Optimized-Root Mean Square Propagation) Classification for Prediction of Polycystic Ovary Syndrome (PCOS) using Hybrid Machine Learning Technique. Int. J. Adv. Comput. Sci. Appl. 2022, 13, 588–596. [Google Scholar]
- Yağmur, S.; Özkurt, N. Convolutional neural network hyperparameter tuning with Adam optimizer for ECG classification. In Proceedings of the 2020 Innovations in Intelligent Systems and Applications Conference (ASYU), Istanbul, Turkey, 15–17 October 2020; IEEE: Piscataway, NJ, USA, 2020. [Google Scholar]
- Gordon, G.J.; Jensen, R.V.; Hsiao, L.L.; Gullans, S.R.; Blumenstock, J.E.; Ramaswamy, S.; William, G.; David, R.; Sugarbaker, J.; Bueno, R. Translation of Microarray Data into Clinically Relevant Cancer Diagnostic Tests Using Gene Expression Ratios in Lung Cancer and Mesothelioma1. Cancer Res. 2002, 62, 4963–4967. [Google Scholar] [PubMed]
- Liu, T.-C.; Kalugin, P.N.; Wilding, J.L.; Bodmer, W.F. GMMchi: Gene expression clustering using Gaussian mixture modeling. BMC Bioinform. 2022, 23, 457. [Google Scholar] [CrossRef]
- Park, C.H.; Park, H. Fingerprint classification using fast Fourier transform and nonlinear discriminant analysis. Pattern Recognit. 2005, 38, 495–503. [Google Scholar] [CrossRef]
- Kim, P.M.; Tidor, B. Subsystem identification through dimensionality reduction of large-scale gene expression data. Genome Res. 2003, 13, 1706–1718. [Google Scholar] [CrossRef]
- Almugren, N.; Alshamlan, H. A survey on hybrid feature selection methods in microarray gene expression data for cancer classification. IEEE Access 2019, 7, 78533–78548. [Google Scholar] [CrossRef]
- Cai, Z.; Xu, D.; Zhang, Q.; Zhang, J.; Ngai, S.-M.; Shao, J. Classification of lung cancer using ensemble-based feature selection and machine learning methods. Mol. Biosyst. 2015, 11, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Alhenawi, E.; Al-Sayyed, R.; Hudaib, A.; Mirjalili, S. Feature selection methods on gene expression microarray data for cancer classification: A systematic review. Comput. Biol. Med. 2022, 140, 105051. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Huo, Y.; Xin, L.; Tian, B.; Yu, B. Feature selection and tumor classification for microarray data using relaxed Lasso and generalized multi-class support vector machine. J. Theor. Biol. 2019, 463, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Dagnew, G.; Shekar, B.H. Ensemble learning-based classification of microarray cancer data on tree-based features. Cogn. Comput. Syst. 2021, 3, 48–60. [Google Scholar] [CrossRef]
- Cui, X.; Li, Y.; Fan, J.; Wang, T.; Zheng, Y. A hybrid improved dragonfly algorithm for feature selection. IEEE Access 2020, 8, 155619–155629. [Google Scholar] [CrossRef]
- Mafarja, M.; Aljarah, I.; Heidari, A.A.; Faris, H.; Fournier-Viger, P.; Li, X.; Mirjalili, S. Binary dragonfly optimization for feature selection using time-varying transfer functions. Knowl.-Based Syst. 2018, 161, 185–204. [Google Scholar] [CrossRef]
- Rahman, C.M.; Rashid, T.A. Dragonfly Algorithm and Its Applications in Applied Science Survey. Comput. Intell. Neurosci. 2019, 2019, 9293617. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, W.; Liu, Y. A hybrid approach for biomarker discovery from microarray gene expression data for cancer classification. Cancer Inform. 2006, 2, 301–311. [Google Scholar] [CrossRef]
- Mohapatra, P.; Chakravarty, S.; Dash, P. Microarray medical data classification using kernel ridge regression and modified cat swarm optimization based gene selection system. Swarm Evol. Comput. 2016, 28, 144–160. [Google Scholar] [CrossRef]
- Huynh, P.H.; Nguyen, V.H.; Do, T.N. A coupling support vector machines with the feature learning of deep convolutional neural networks for classifying microarray gene expression data. In Modern Approaches for Intelligent Information and Database Systems; Springer: Berlin/Heidelberg, Germany, 2018; pp. 233–243. [Google Scholar]
- Dai, W.; Chuang, Y.-Y.; Lu, C.-J. A clustering-based sales forecasting scheme using support vector regression for computer server. Procedia Manuf. 2015, 2, 82–86. [Google Scholar] [CrossRef]
- Kelemen, A.; Zhou, H.; Lawhead, P.; Liang, Y. Naive Bayesian classifier for microarray data. In Proceedings of the 2003 International Joint Conference on Neural Networks, Portland, OR, USA, 20–24 July 2003; Volume 3, pp. 1769–1773. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J.H.; Friedman, J.H. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer: New York, NY, USA, 2009; Volume 2. [Google Scholar]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning; Springer: New York, NY, USA, 2013; Volume 112. [Google Scholar]
- El Kafrawy, P.; Fathi, H.; Qaraad, M.; Kelany, A.K.; Chen, X. An efficient SVM-based feature selection model for cancer classification using high-dimensional microarray data. IEEE Access 2021, 9, 155353–155369. [Google Scholar] [CrossRef]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Xiong, Z.; Cui, Y.; Liu, Z.; Zhao, Y.; Hu, M.; Hu, J. Evaluating explorative prediction power of machine learning algorithms for materials discovery using k-fold forward cross-validation. Comput. Mater. Sci. 2020, 171, 109203. [Google Scholar] [CrossRef]
- Muhajir, D.; Akbar, M.; Bagaskara, A.; Vinarti, R. Improving classification algorithm on education dataset using hyperparameter tuning. Procedia Comput. Sci. 2022, 197, 538–544. [Google Scholar] [CrossRef]
- Elgeldawi, E.; Sayed, A.; Galal, A.R.; Zaki, A.M. Hyperparameter tuning for machine learning algorithms used for arabic sentiment analysis. Informatics 2021, 8, 79. [Google Scholar] [CrossRef]
- Kaur, S.; Aggarwal, H.; Rani, R. Hyper-parameter optimization of deep learning model for prediction of Parkinson’s disease. Mach. Vis. Appl. 2020, 31, 32. [Google Scholar] [CrossRef]
- Masud, M.; Hossain, M.S.; Alhumyani, H.; Alshamrani, S.S.; Cheikhrouhou, O.; Ibrahim, S.; Muhammad, G.; Rashed, A.E.E.; Gupta, B.B. Pre-trained convolutional neural networks for breast cancer detection using ultrasound images. ACM Trans. Internet Technol. 2021, 21, 85. [Google Scholar] [CrossRef]
- Fathi, H.; AlSalman, H.; Gumaei, A.; Manhrawy, I.I.M.; Hussien, A.G.; El-Kafrawy, P. An efficient cancer classification model using microarray and high-dimensional data. Comput. Intell. Neurosci. 2021, 2021, 7231126. [Google Scholar] [CrossRef]
- Guan, P.; Huang, D.; He, M.; Zhou, B. Lung cancer gene expression database analysis incorporating prior knowledge with support vector machine-based classification method. J. Exp. Clin. Cancer Res. 2009, 28, 103–107. [Google Scholar] [CrossRef]
- Gupta, S.; Gupta, M.K.; Shabaz, M.; Sharma, A. Deep learning techniques for cancer classification using microarray gene expression data. Front. Physiol. 2022, 13, 952709. [Google Scholar] [CrossRef]
- Mramor, M.; Leban, G.; Demšar, J.; Zupan, B. Visualization-based cancer microarray data classification analysis. Bioinformatics 2007, 23, 2147–2154. [Google Scholar] [CrossRef]
- Ke, L.; Li, M.; Wang, L.; Deng, S.; Ye, J.; Yu, X. Improved swarm-optimization-based filter-wrapper gene selection from microarray data for gene expression tumor classification. Pattern Anal. Appl. 2022, 26, 455–472. [Google Scholar] [CrossRef]
- Xia, D.; Leon, A.J.; Cabanero, M.; Pugh, T.J.; Tsao, M.S.; Rath, P.; Siu, L.L.-Y.; Yu, C.; Bedard, P.L.; Shepherd, F.A.; et al. Minimalist approaches to cancer tissue-of-origin classification by DNA methylation. Mod. Pathol. 2020, 33, 1874–1888. [Google Scholar] [CrossRef]
- Morani, F.; Bisceglia, L.; Rosini, G.; Mutti, L.; Melaiu, O.; Landi, S.; Gemignani, F. Identification of overexpressed genes in malignant pleural mesothelioma. Int. J. Mol. Sci. 2021, 22, 2738. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl.No | Statistical Features | Mixture Model | FFT | ||
|---|---|---|---|---|---|
| Adeno Carcinoma | Meso Cancer | Adeno Carcinoma | Meso Cancer | ||
| 1 | Mean | 12.77239 | 84.4254 | 50,051.74 | 64,399.1406 |
| 2 | Variance | 28,701.74 | 72,406.87 | 8.14 × 108 | 1,207,801,420 |
| 3 | Skewness | 25.62594 | 11.83928 | 22.08858 | 17.9010876 |
| 4 | Kurtosis | 1008.477 | 211.3989 | 1392.65 | 1072.04601 |
| 5 | PCC | 0.84004 | 0.926835 | 0.944664 | 0.94001594 |
| 6 | t-test | 0.017655 | 3.14 × 10−18 | 2.06 × 10−24 | 1.096 × 10−21 |
| 7 | p-value < 0.01 | 0.493103 | 0.5 | 0.5 | 0.5 |
| 8 | Canonical Correlation Analysis (CCA) | 0.3852 | 0.3371 | ||
| Classifiers | Mixture Model DimRe Method and without FS | FFT DimRe Method and without FS | Mixture Model DimRe Method and with DF FS | FFT DimRe Method and with DF FS | ||||
|---|---|---|---|---|---|---|---|---|
| Training MSE | Testing MSE | Training MSE | Testing MSE | Training MSE | Testing MSE | Training MSE | Testing MSE | |
| Nonlinear Regression | 3.84 × 10−7 | 5.63 × 10−5 | 3.11 × 10−6 | 0.000016 | 1.44 × 10−6 | 3.6 × 10−6 | 2.54 × 10−9 | 6.24 × 10−8 |
| Naïve Bayesian | 1.56 × 10−9 | 2.93 × 10−7 | 5.61 × 10−9 | 3.24 × 10−8 | 3.48 × 10−6 | 4.2 × 10−5 | 3.03 × 10−7 | 5.04 × 10−5 |
| Random Forest | 1.23 × 10−8 | 1.94 × 10−5 | 1.44 × 10−7 | 6.89 × 10−5 | 3.06 × 10−7 | 5.76 × 10−6 | 6.4 × 10−6 | 2.92 × 10−5 |
| Decision Tree | 3.25 × 10−6 | 5.48 × 10−5 | 2.56 × 10−6 | 4.49 × 10−5 | 2.89 × 10−7 | 2.6 × 10−5 | 8.1 × 10−7 | 4.76 × 10−5 |
| SVM(RBF) | 2.6 × 10−8 | 1.69 × 10−6 | 8.1 × 10−8 | 2.5 × 10−7 | 1.96 × 10−9 | 5.18 × 10−7 | 1.02 × 10−8 | 1.56 × 10−7 |
| Classifier | Parameter Value |
|---|---|
| NR | T1 = 0.85, T2 = 0.65, n1, n2, and n3 is retrieved from (15), b0 = 0.01, Convergence Criteria (ConvCrit) = MSE |
| NB | Smoothing parameter, α = 0.06, Prior Probability = 0.15, ConvCrit = MSE |
| RF | Number of trees NT = 100, Depth D = 10, ConvCrit = MSE |
| DT | Depth D = 10, ConvCrit = MSE |
| SVM (RBF) | Width of the radial basis function, = 1, ConvCrit = MSE |
| Truth of Clinical Situation | Observed | ||
|---|---|---|---|
| Adeno | Meso | ||
| Actual | Adeno | TP | FN |
| Meso | FP | TN | |
| Performance Metrics | Derived from Confusion Matrix |
|---|---|
| Accuracy | |
| F1 Score | |
| Mathews Correlation Coefficient | |
| Error Rate | |
| Youden Index | |
| Kappa | )/(1-Eacc) |
| DimRe Method | Mixture Model | FFT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classifiers | NR | NB | RF | DT | SVM (RBF) | NR | NB | RF | DT | SVM (RBF) |
| Parameters | ||||||||||
| Accuracy | 67.403 | 76.243 | 75.691 | 65.746 | 59.669 | 72.928 | 88.950 | 62.983 | 54.144 | 60.221 |
| F1 Score | 78.067 | 84.912 | 84.397 | 76.692 | 70.445 | 81.369 | 93.464 | 74.131 | 66.122 | 69.492 |
| MCC | 0.197 | 0.307 | 0.317 | 0.179 | 0.194 | 0.404 | 0.583 | 0.170 | 0.067 | 0.315 |
| Error Rate | 32.597 | 23.757 | 24.309 | 34.254 | 40.331 | 27.072 | 11.050 | 37.017 | 45.856 | 39.779 |
| Youden Index | 24.839 | 35.505 | 37.398 | 22.839 | 25.742 | 51.978 | 53.398 | 22.065 | 8.839 | 41.763 |
| Kappa | 0.178 | 0.298 | 0.304 | 0.159 | 0.153 | 0.353 | 0.578 | 0.145 | 0.052 | 0.230 |
| DimRe Method | Mixture Model | FFT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classifiers | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) |
| Parameters | ||||||||||
| Accuracy | 67.956 | 68.508 | 53.039 | 60.221 | 91.160 | 85.083 | 58.011 | 53.591 | 67.956 | 82.873 |
| F1 Score | 77.863 | 78.967 | 65.021 | 71.875 | 94.558 | 90.970 | 68.333 | 65.854 | 78.519 | 88.889 |
| MCC | 0.277 | 0.209 | 0.057 | 0.124 | 0.715 | 0.481 | 0.217 | 0.042 | 0.203 | 0.554 |
| Error Rate | 32.044 | 31.492 | 46.961 | 39.779 | 8.840 | 14.917 | 41.989 | 46.409 | 32.044 | 17.127 |
| Youden Index | 35.742 | 26.172 | 7.505 | 16.172 | 76.538 | 48.731 | 28.860 | 5.613 | 25.505 | 66.538 |
| Kappa | 0.240 | 0.191 | 0.043 | 0.103 | 0.711 | 0.481 | 0.163 | 0.033 | 0.184 | 0.524 |
| Classifiers | Optimal Values | Initial Values | |||||
|---|---|---|---|---|---|---|---|
| β1 | β2 | ||||||
| NR | 0.5 | 0.5 | 0.2 | 0.28 | 0.42 | 0.1 | 0.15 |
| NB | 0.6 | 0.4 | 0.26 | 0.32 | 0.5 | 0.1 | 0.2 |
| RF | 0.45 | 0.55 | 0.38 | 0.4 | 0.38 | 0.1 | 0.25 |
| DT | 0.55 | 0.45 | 0.33 | 0.41 | 0.6 | 0.15 | 0.2 |
| SVM(RBF) | 0.35 | 0.65 | 0.32 | 0.45 | 0.5 | 0.1 | 0.2 |
| Classifiers with Adam Hyper-Parameter Tuning | Mixture Model DimRe Method and with DF FS | FFT DimRe Method and with DF FS | ||
|---|---|---|---|---|
| Training Accuracy | Testing Accuracy | Training Accuracy | Testing Accuracy | |
| Nonlinear Regression | 90.31 | 88.23 | 91.34 | 89.84 |
| Naïve Bayesian | 91.23 | 89.29 | 92.56 | 90.39 |
| Random Forest | 92.97 | 91.84 | 93.47 | 91.95 |
| Decision Tree | 86.31 | 82.87 | 92.54 | 90.39 |
| SVM (RBF) | 98.66 | 96.47 | 93.79 | 90.84 |
| Classifiers with RanAdam Hyper-parameter Tuning | Mixture Model DimRe Method and with DF FS | FFT DimRe Method and with DF FS | ||
|---|---|---|---|---|
| Training Accuracy | Testing Accuracy | Training Accuracy | Testing Accuracy | |
| Nonlinear Regression | 92.62 | 89.74 | 92.44 | 90.64 |
| Naïve Bayesian | 95.87 | 93.22 | 93.52 | 90.51 |
| Random Forest | 94.25 | 92.86 | 94.62 | 92.19 |
| Decision Tree | 92.37 | 90.219 | 95.61 | 93.53 |
| SVM (RBF) | 93.66 | 90.72 | 99.41 | 98.86 |
| DimRe Method | Mixture Model | FFT Method | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classifiers | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) |
| Parameters | ||||||||||
| Accuracy | 80.110 | 87.293 | 87.845 | 82.873 | 94.475 | 87.845 | 88.398 | 88.950 | 88.398 | 87.845 |
| F1 Score | 87.413 | 92.256 | 92.667 | 89.199 | 96.667 | 92.466 | 92.929 | 93.243 | 93.023 | 92.414 |
| MCC | 0.417 | 0.570 | 0.572 | 0.494 | 0.805 | 0.618 | 0.607 | 0.631 | 0.586 | 0.630 |
| Error Rate | 19.890 | 12.707 | 12.155 | 17.127 | 5.525 | 12.155 | 11.602 | 11.050 | 11.602 | 12.155 |
| Youden Index | 47.849 | 59.075 | 57.183 | 56.301 | 80.538 | 67.419 | 62.968 | 66.194 | 57.849 | 69.978 |
| Kappa | 0.406 | 0.569 | 0.572 | 0.483 | 0.805 | 0.612 | 0.606 | 0.630 | 0.586 | 0.620 |
| DimRe Method | Mixture Model | FFT Method | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classifiers | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) | Nonlinear Regression | Naïve Bayesian | Random Forest | Decision Tree | SVM (RBF) |
| Parameters | ||||||||||
| Accuracy | 86.740 | 91.160 | 91.160 | 88.398 | 87.293 | 88.950 | 85.635 | 88.398 | 90.608 | 98.343 |
| F1 Score | 91.892 | 94.667 | 94.702 | 93.023 | 92.256 | 93.289 | 91.216 | 93.069 | 94.352 | 98.997 |
| MCC | 0.557 | 0.689 | 0.681 | 0.586 | 0.570 | 0.621 | 0.520 | 0.576 | 0.665 | 0.943 |
| Error Rate | 13.260 | 8.840 | 8.840 | 11.602 | 12.707 | 11.050 | 14.365 | 11.602 | 9.392 | 1.657 |
| Youden Index | 58.409 | 68.860 | 66.301 | 57.849 | 59.075 | 63.634 | 54.516 | 55.290 | 65.634 | 95.441 |
| Kappa | 0.556 | 0.689 | 0.680 | 0.586 | 0.569 | 0.620 | 0.519 | 0.575 | 0.665 | 0.942 |
| Classifiers | Mixture Model DimRe Method and with DF FS | FFT DimRe Method and with DF FS | ||
|---|---|---|---|---|
| Accuracy Improvement by Adam Method (%) | Accuracy Improvement by RanAdam Method (%) | Accuracy Improvement by Adam Method (%) | Accuracy Improvement by RanAdam Method (%) | |
| Nonlinear Regression | 15.172 | 21.65 | 3.145 | 4.347 |
| Naïve Bayesian | 21.519 | 24.84 | 34.375 | 32.258 |
| Random Forest | 39.623 | 41.81 | 39.752 | 39.375 |
| Decision Tree | 27.333 | 31.875 | 23.125 | 25 |
| SVM(RBF) | 3.509 | 4.43 | 5.66 | 15.73 |
| Classifiers | Without FS | With DF FS | With DF FS and Adam Tuning | With DF FS and RanAdam Tuning |
|---|---|---|---|---|
| Nonlinear Regression | O (2n3 log2n) | O (2n6 log 2n) | O (2n6 log 2n) | O (2n4 log2n) |
| Naïve Bayesian | O (2n4 log2n) | O (2n7 log 2n) | O (2n7 log 2n) | O (2n5log2n) |
| Random Forest | O (2n3 log2n) | O (2n6 log 2n) | O (2n6 log 2n) | O (2n4 log2n) |
| Decision Tree | O (2n3 log2n) | O (2n6 log 2n) | O (2n6 log 2n) | O (2n4 log2n) |
| SVM(RBF) | O (2n2 log4n) | O (2n5 log 4n) | O (2n5 log 4n) | O (2n3 log4n) |
| S.No | Author (with Year) | Database | Classifier | Classes | Performance Accuracy in% |
|---|---|---|---|---|---|
| 1 | Azzawi (2015) [31] | National Library of Medicine and Kent Ridge Bio-medical Dataset | SVM, MLP, RBFN | Adenocarcinoma, Meso | 91.39 91.72 89.82 |
| 2 | Gordon (2002) [39] | Gordon MAGE Data | MAGE ratios | Adenocarcinoma, Meso | 90 |
| 3 | Fathi et al. (2021) [65] | Gordon MAGE Data | Decision Tree with feature fusion | Adenocarcinoma, Meso | 85 |
| 4 | Guan et al. (2009) [66] | Affymetrix Human GeneAtlas U95Av2 microarray dataset | SVM (RBF) with gene based feature | Adenocarcinoma, Meso | 94 |
| 5 | Gupta et al. (2022) [67] | TCGA dataset | Deep CNN | Adenocarcinoma, Meso | 92 |
| 6 | Mramor et al. (2007) [68] | Gordon MAGE Data | SVM, Naïve Bayes, KNN, Decision Tree | Adenocarcinoma, Meso | 94.67 90.35 75.28 91.21 |
| 7 | Lin Ke (2022) [69] | Gordon MAGE Data | DT—C4.5 | Adenocarcinoma, Meso | 93 |
| 8 | Daniel Xia et al. (2020) [70] | Gordon MAGE Data | Minimalist Cancer Classifier | Adenocarcinoma, Meso | 90.6 |
| 9 | Morani et al.(2021) [71] | TCGA and GEO Dataset | Multivariate cox regression analysis | Adenocarcinoma, Meso | 90 |
| 10 | This Research | Gordon MAGE Data | RanAdam Hyper-parameter tuning for FFT DimRe techniques with DF FS and SVM (RBF) Classification | Adenocarcinoma, Meso | 98.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
M S, K.; Rajaguru, H.; Nair, A.R. Enhancement of Classifier Performance with Adam and RanAdam Hyper-Parameter Tuning for Lung Cancer Detection from Microarray Data—In Pursuit of Precision. Bioengineering 2024, 11, 314. https://doi.org/10.3390/bioengineering11040314
M S K, Rajaguru H, Nair AR. Enhancement of Classifier Performance with Adam and RanAdam Hyper-Parameter Tuning for Lung Cancer Detection from Microarray Data—In Pursuit of Precision. Bioengineering. 2024; 11(4):314. https://doi.org/10.3390/bioengineering11040314
Chicago/Turabian StyleM S, Karthika, Harikumar Rajaguru, and Ajin R. Nair. 2024. "Enhancement of Classifier Performance with Adam and RanAdam Hyper-Parameter Tuning for Lung Cancer Detection from Microarray Data—In Pursuit of Precision" Bioengineering 11, no. 4: 314. https://doi.org/10.3390/bioengineering11040314
APA StyleM S, K., Rajaguru, H., & Nair, A. R. (2024). Enhancement of Classifier Performance with Adam and RanAdam Hyper-Parameter Tuning for Lung Cancer Detection from Microarray Data—In Pursuit of Precision. Bioengineering, 11(4), 314. https://doi.org/10.3390/bioengineering11040314