

Contractile and Genetic Characterization of Cardiac Constructs Engineered from Human Induced Pluripotent Stem Cells: Modeling of Tuberous Sclerosis Complex and the Effects of Rapamycin

 ,
, 
Abstract
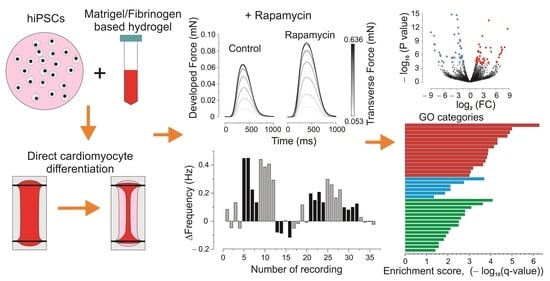
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
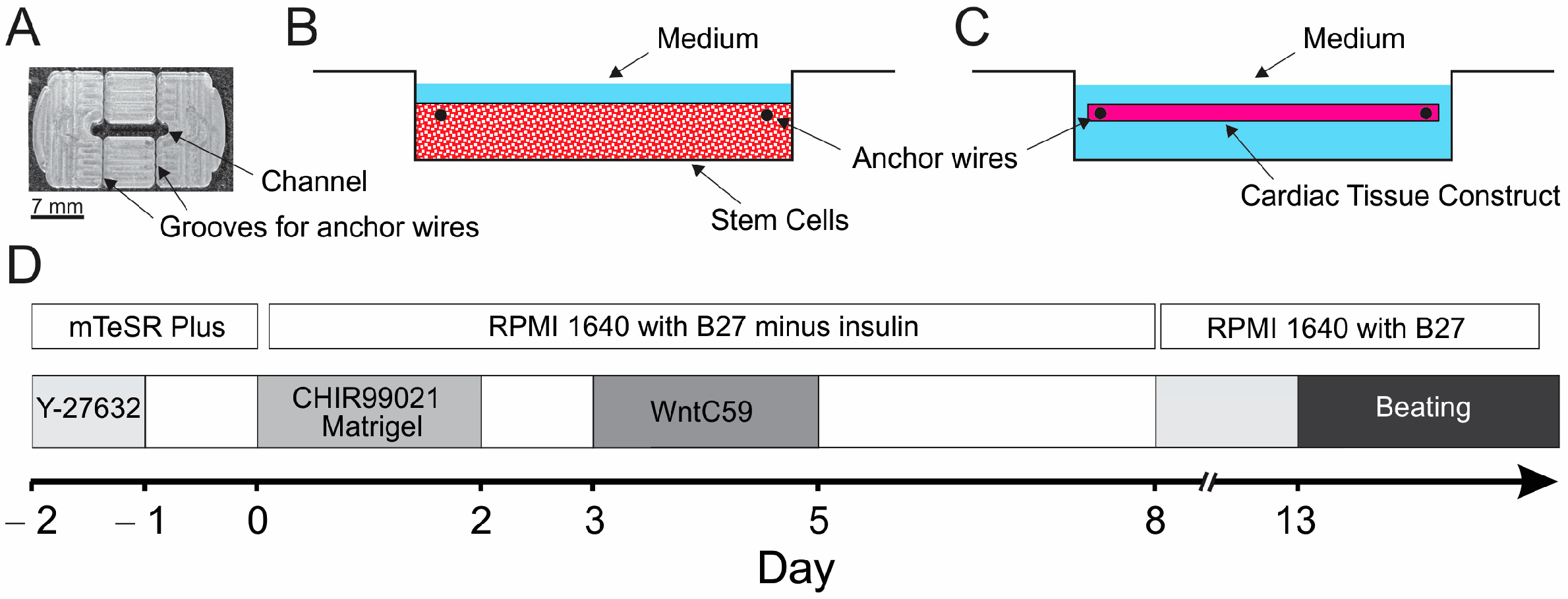
2.1. Preparation of PDMS Casting Mold and Cell Mixture
2.2. Derivation and Validation of hiPSCs
2.3. hiPSC Differentiation
2.4. Data Registration and Processing
2.5. RNA Preparation, Next-Generation Sequencing (NGS), and Gene Set Enrichment Analysis (GSEA)
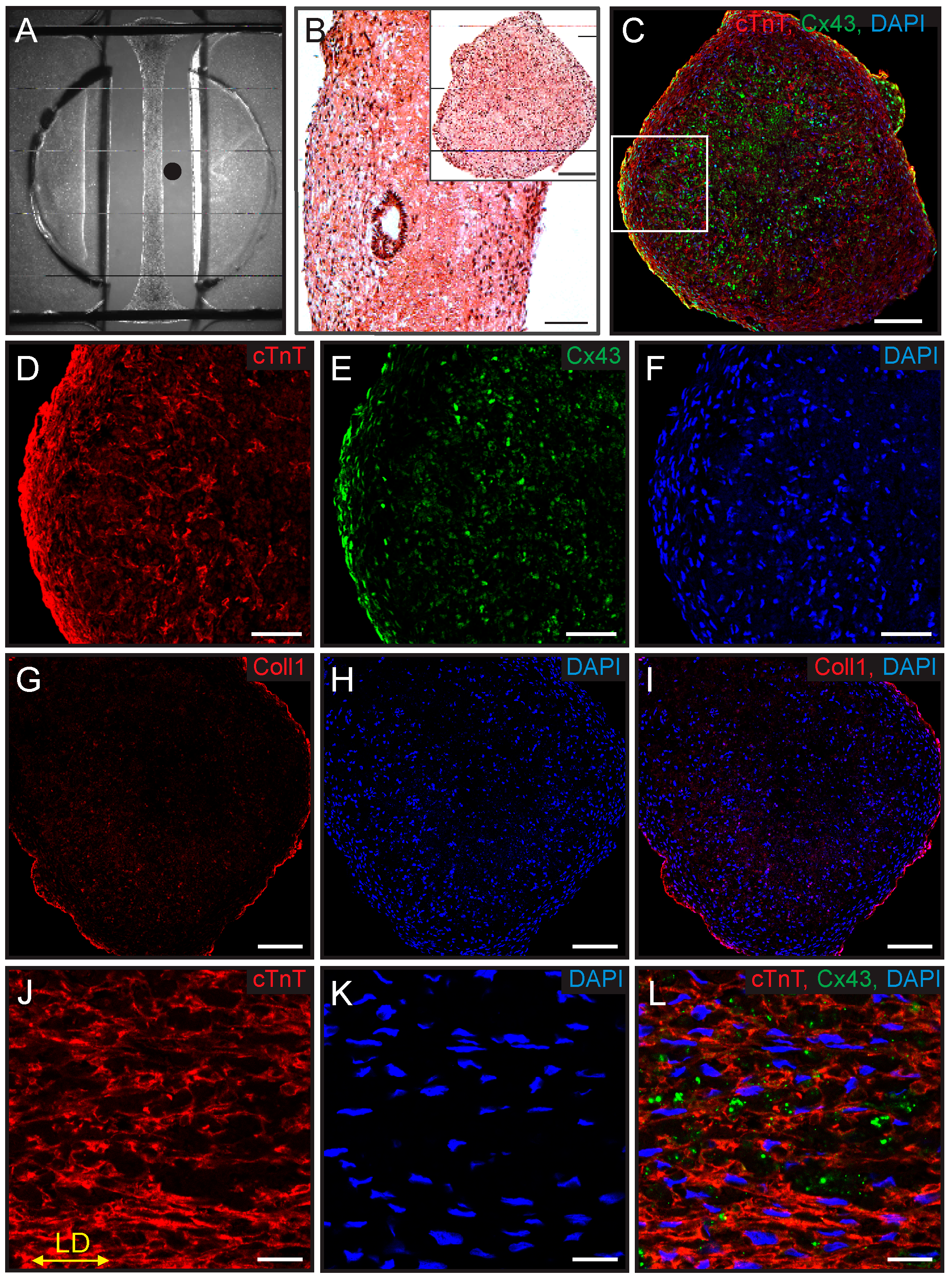
2.6. Immunostaining and Histology
2.7. Descriptive Statistics
3. Results
3.1. Growth and Characterization of ECTCs
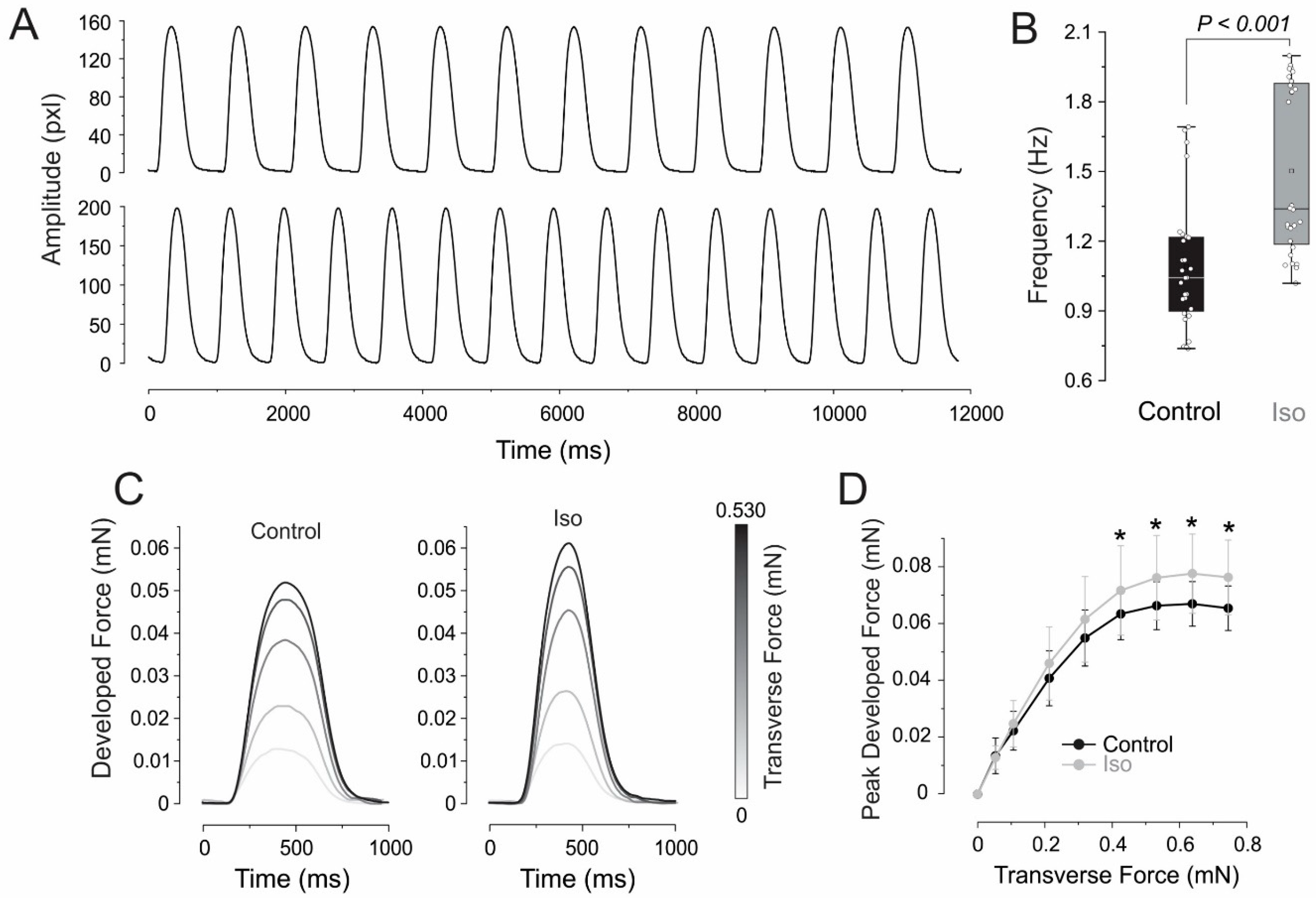
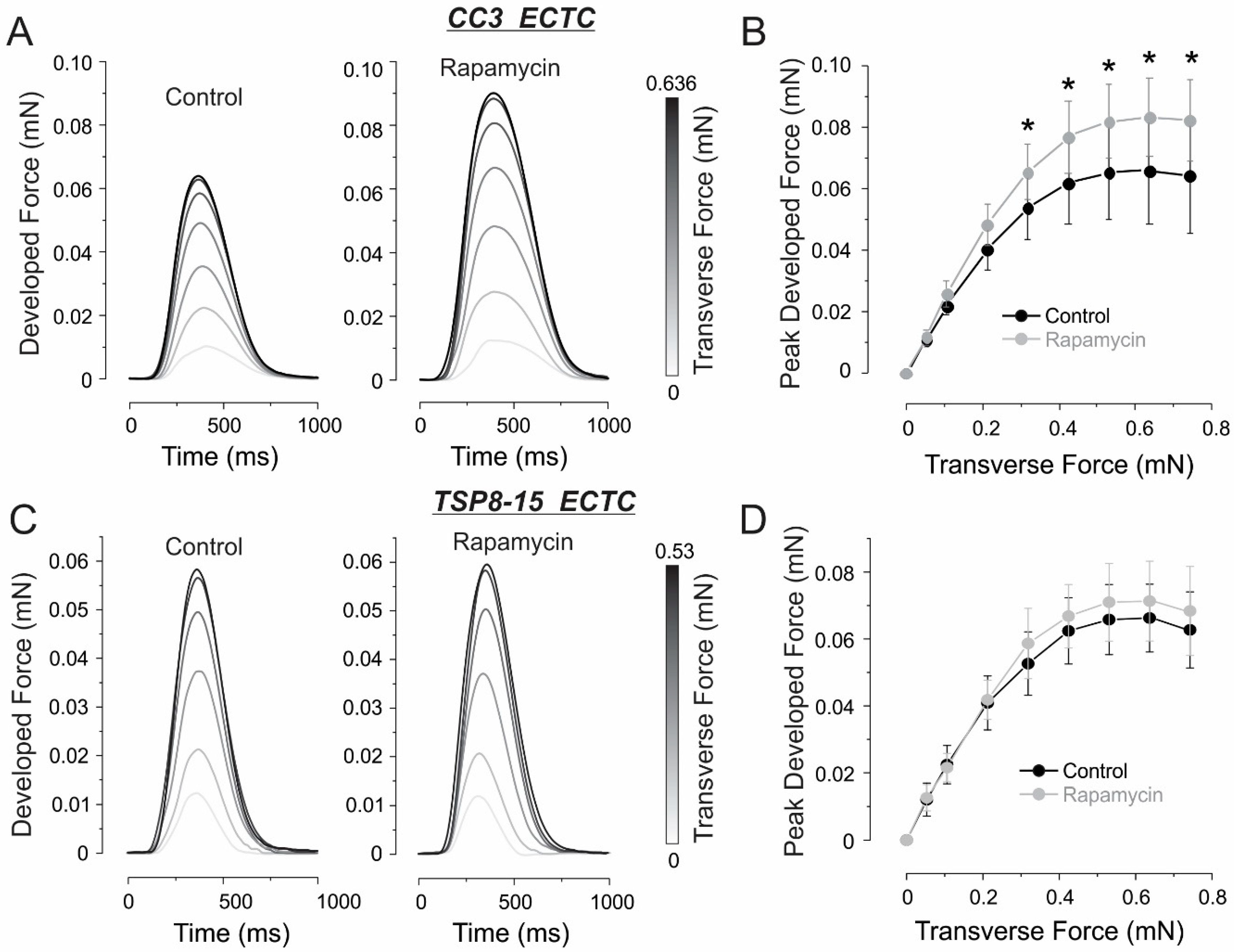
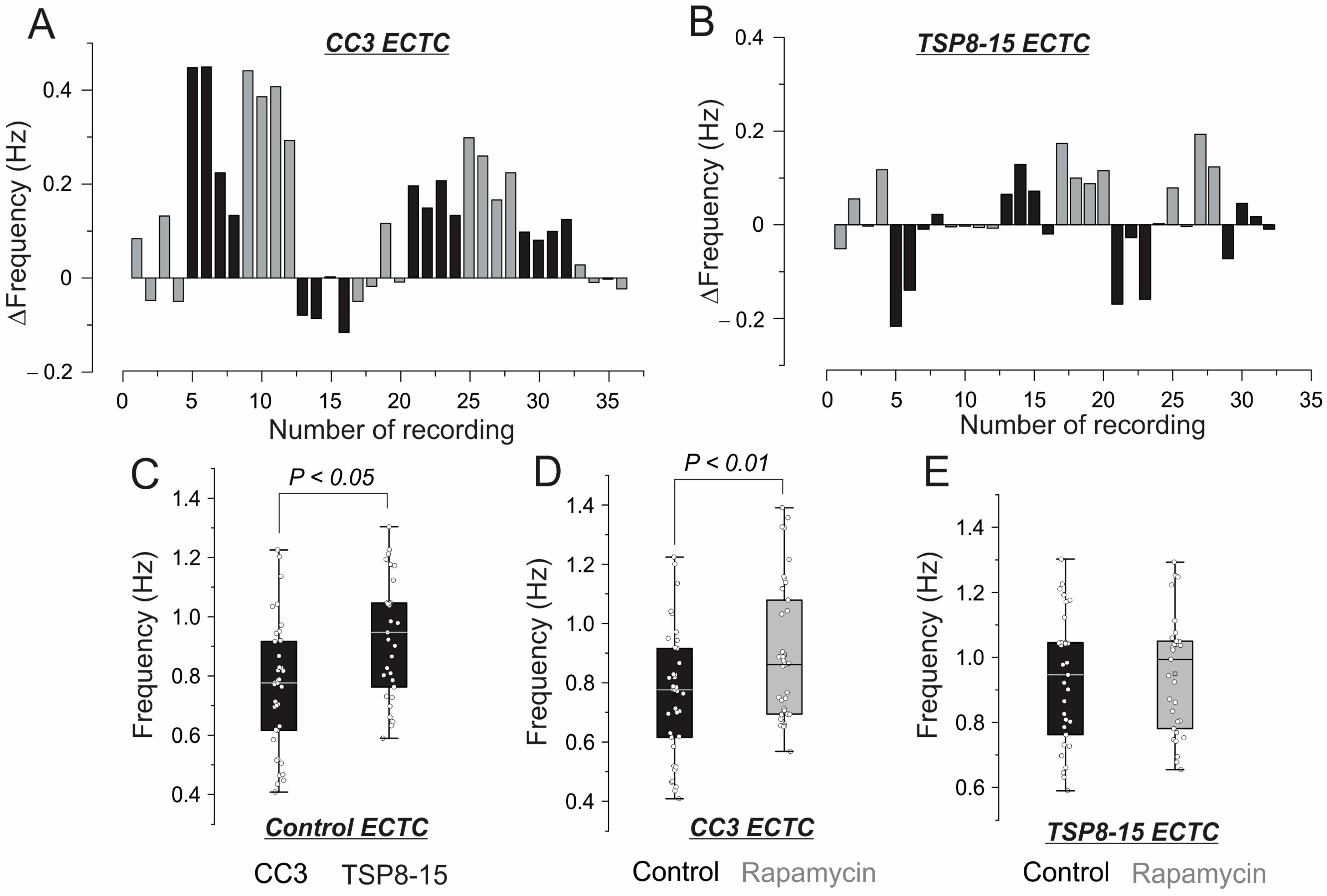
3.2. Inotropic and Chronotropic Effects of Rapamycin in CC3 and TSC Constructs
3.3. Elastic Properties
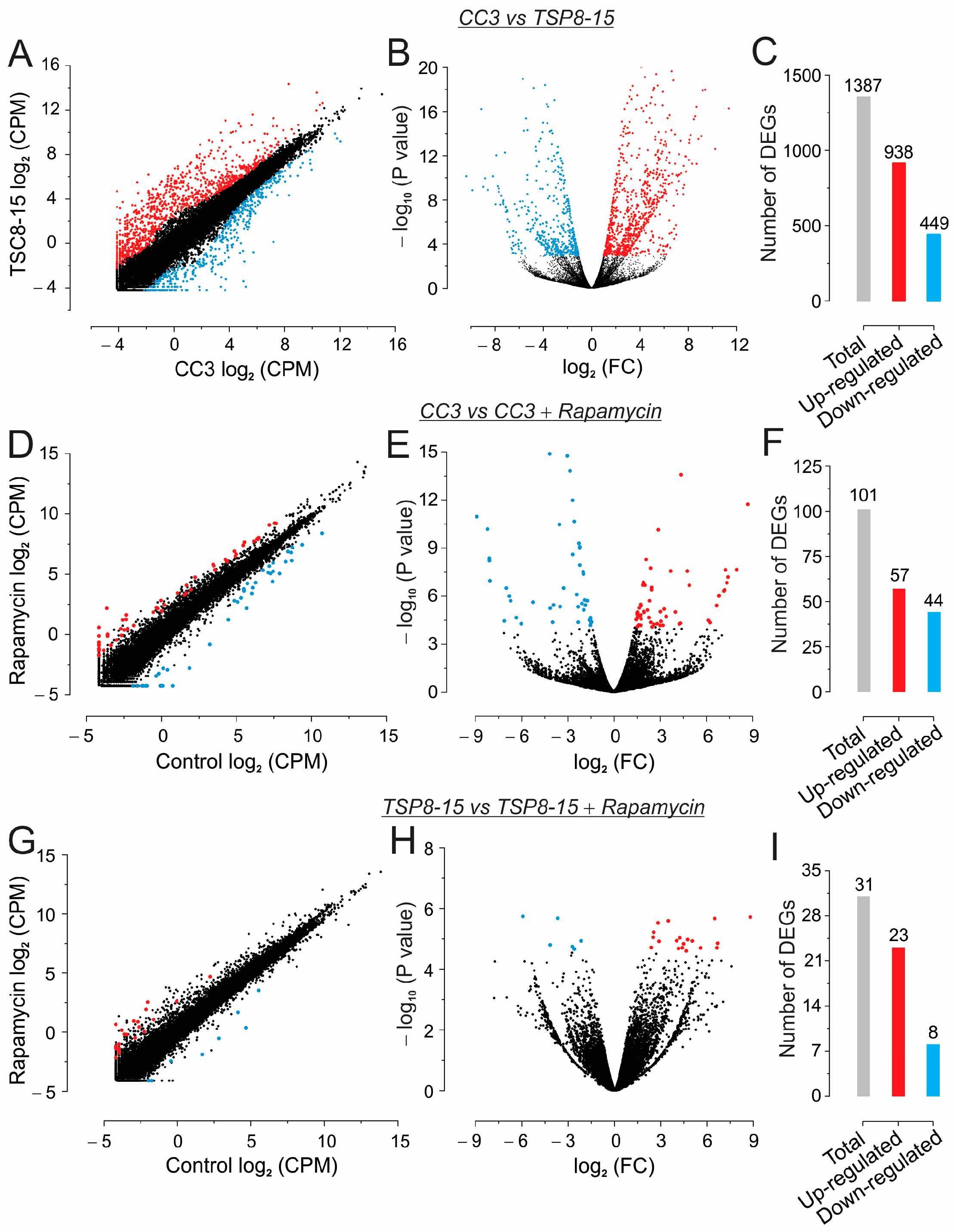
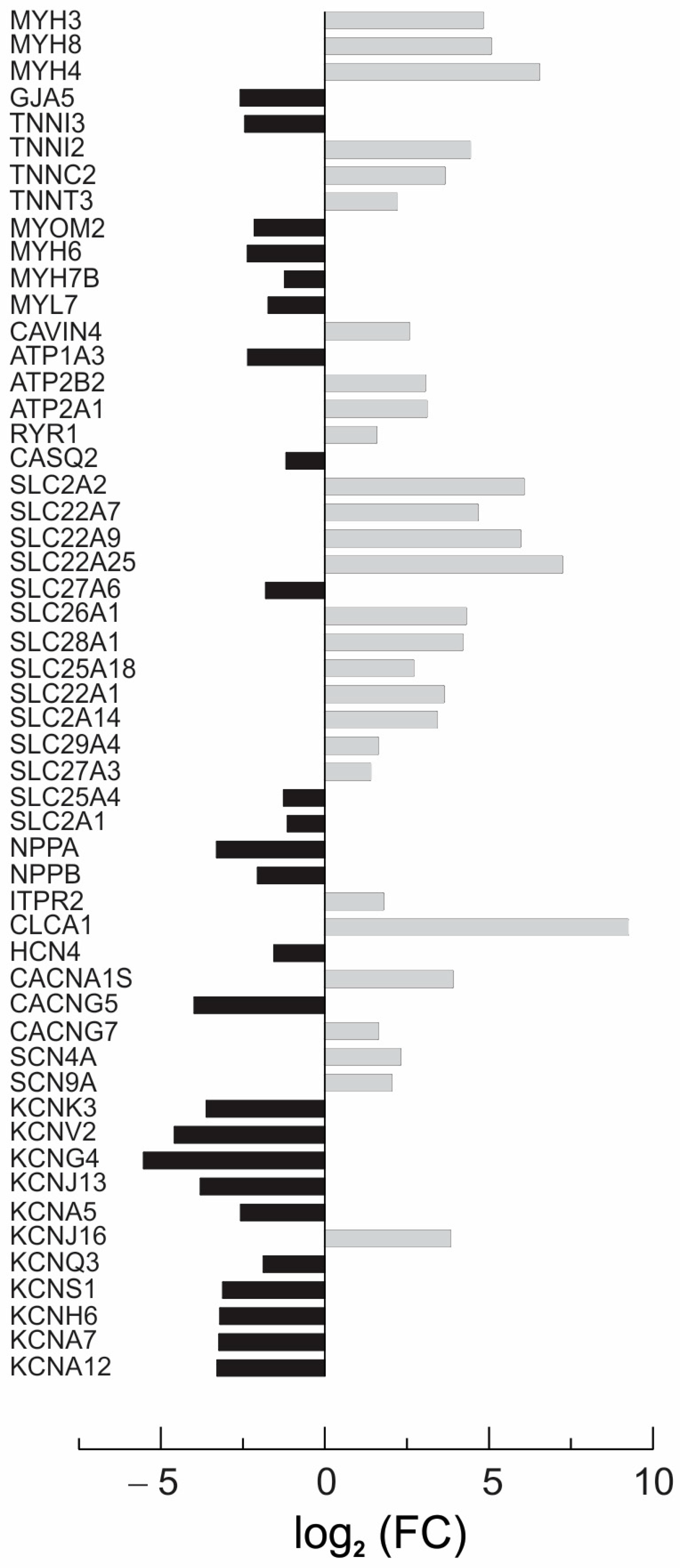
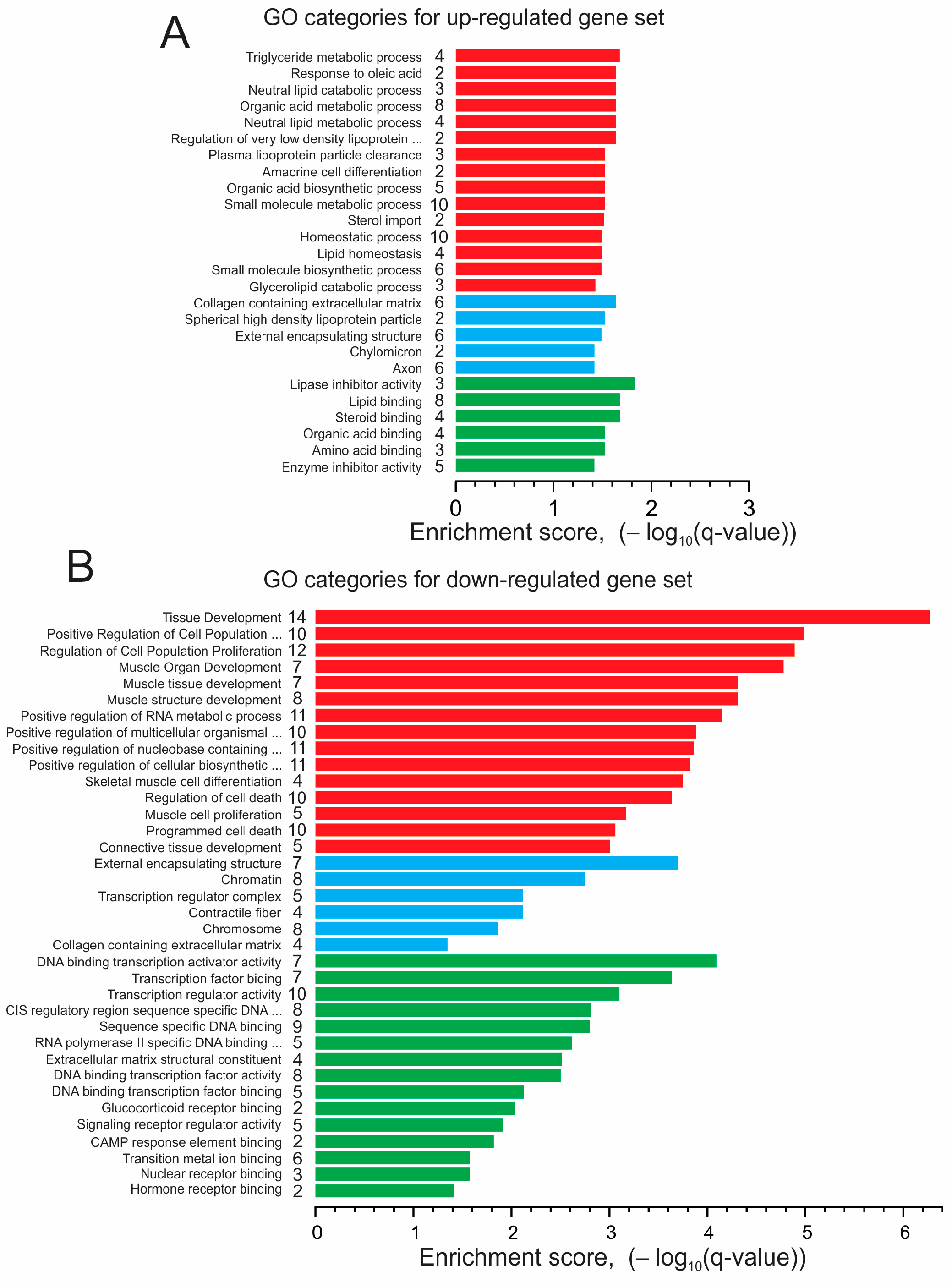
3.4. Next-Generation Sequencing (NGS) and Gene Ontology Enrichment Analysis of Differentially Expressed Genes (DEGs)
4. Discussion
4.1. Growing ECTCs
4.2. Inotropic Effect of Rapamycin in CC3 ECTCs
4.3. Resistance to Rapamycin in TSP8-15 ECTCs
4.4. Effect of Rapamycin on CC3 ECTC Gene Expression
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, D.E.; Hunziker, R.; Wikswo, J.P. Fitting tissue chips and microphysiological systems into the grand scheme of medicine, biology, pharmacology, and toxicology. Exp. Biol. Med. 2017, 242, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Fukuda, K. Methods of induced pluripotent stem cells for clinical application. World J. Stem Cells 2015, 7, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Fujita, J.; Fujita, C.; Yamaguchi, M.; Kanaami, S.; Ohno, R.; Sakamoto, K.; Kodama, M.; Kurokawa, J.; Kanazawa, H.; et al. Efficient Large-Scale 2D Culture System for Human Induced Pluripotent Stem Cells and Differentiated Cardiomyocytes. Stem Cell Rep. 2017, 9, 1406–1414. [Google Scholar] [CrossRef]
- Orlova, K.A.; Crino, P.B. The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 2010, 1184, 87–105. [Google Scholar] [CrossRef]
- Mühler, E.G.; Turniski-Harder, V.; Engelhardt, W.; von Bernuth, G. Cardiac involvement in tuberous sclerosis. Heart 1994, 72, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, P.; Babu, J.S.; Al-Bulushi, A. Right atrial rhabdomyoma acting as the substrate for Wolff-Parkinson-White syndrome in a 3-month-old infant. Acta Cardiol. 2005, 60, 543–545. [Google Scholar] [CrossRef]
- Hinton, R.B.; Prakash, A.; Romp, R.L.; Krueger, D.A.; Knilans, T.K. Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group. J. Am. Heart Assoc. 2014, 3, e001493. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Minowa, O.; Sugitani, Y.; Takai, S.; Mitani, H.; Kobayashi, E.; Noda, T.; Hino, O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8762–8767. [Google Scholar] [CrossRef]
- Zeng, L.H.; Xu, L.; Gutmann, D.H.; Wong, M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann. Neurol. 2008, 63, 444–453. [Google Scholar] [CrossRef]
- Meikle, L.; McMullen, J.R.; Sherwood, M.C.; Lader, A.S.; Walker, V.; Chan, J.A.; Kwiatkowski, D.J. A mouse model of cardiac rhabdomyoma generated by loss of Tsc1 in ventricular myocytes. Hum. Mol. Genet. 2005, 14, 429–435. [Google Scholar] [CrossRef]
- Krueger, D.A.; Sadhwani, A.; Byars, A.W.; de Vries, P.J.; Franz, D.N.; Whittemore, V.H.; Filip-Dhima, R.; Murray, D.; Kapur, K.; Sahin, M. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 2017, 4, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Overwater, I.E.; Rietman, A.B.; Mous, S.E.; Bindels-de Heus, K.; Rizopoulos, D.; Ten Hoopen, L.W.; van der Vaart, T.; Jansen, F.E.; Elgersma, Y.; Moll, H.A.; et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 2019, 93, e200–e209. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N.; Baker, H.; Vézina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Kuo, C.J.; Crabtree, G.R.; Blenis, J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 1992, 69, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a025924. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef]
- Cheng, Y.; Felix, B.; Othmer, H.G. The Roles of Signaling in Cytoskeletal Changes, Random Movement, Direction-Sensing and Polarization of Eukaryotic Cells. Cells 2020, 9, 1437. [Google Scholar] [CrossRef]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Rapamycin Inhibits Cardiac Hypertrophy by Promoting Autophagy via the MEK/ERK/Beclin-1 Pathway. Front. Physiol. 2016, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Chen, W.; Yan, M.; Liu, J.; Luo, H.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020, 45, 195–209. [Google Scholar] [CrossRef]
- Wang, J.; Maimaitili, Y.; Zheng, H.; Yu, J.; Guo, H.; Ma, H.P.; Chen, C.L. The influence of rapamycin on the early cardioprotective effect of hypoxic preconditioning on cardiomyocytes. Arch. Med. Sci. 2017, 13, 947–955. [Google Scholar] [CrossRef]
- Kurdi, A.; De Meyer, G.R.; Martinet, W. Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1267–1279. [Google Scholar] [CrossRef]
- Cai, Z.; He, Y.; Chen, Y. Role of Mammalian Target of Rapamycin in Atherosclerosis. Curr. Mol. Med. 2018, 18, 216–232. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sukhorukov, V.N.; Zhuravlev, A.; Orekhov, N.A.; Kalmykov, V.; Orekhov, A.N. Modulating mTOR Signaling as a Promising Therapeutic Strategy for Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1153. [Google Scholar] [CrossRef]
- Hirt, M.N.; Hansen, A.; Eschenhagen, T. Cardiac tissue engineering: State of the art. Circ. Res. 2014, 114, 354–367. [Google Scholar] [CrossRef]
- Dou, W.; Malhi, M.; Zhao, Q.; Wang, L.; Huang, Z.; Law, J.; Liu, N.; Simmons, C.A.; Maynes, J.T.; Sun, Y. Microengineered platforms for characterizing the contractile function of in vitro cardiac models. Microsyst. Nanoeng. 2022, 8, 26. [Google Scholar] [CrossRef]
- Sidorov, V.Y.; Samson, P.C.; Sidorova, T.N.; Davidson, J.M.; Lim, C.C.; Wikswo, J.P. I-Wire Heart-on-a-Chip I: Three-dimensional cardiac tissue constructs for physiology and pharmacology. Acta Biomater. 2017, 48, 68–78. [Google Scholar] [CrossRef]
- Schroer, A.K.; Shotwell, M.S.; Sidorov, V.Y.; Wikswo, J.P.; Merryman, W.D. I-Wire Heart-on-a-Chip II: Biomechanical analysis of contractile, three-dimensional cardiomyocyte tissue constructs. Acta Biomater. 2017, 48, 79–87. [Google Scholar] [CrossRef]
- Sulgin, A.A.; Sidorova, T.N.; Sidorov, V.Y. Growth and characterization of a tissue-engineered construct from human coronary artery smooth muscle cells. Biulleten Sib. Meditsiny 2020, 19, 85–95. [Google Scholar] [CrossRef]
- Pino, J.C.; Lubbock, A.L.R.; Harris, L.A.; Gutierrez, D.B.; Farrow, M.A.; Muszynski, N.; Tsui, T.; Sherrod, S.D.; Norris, J.L.; McLean, J.A.; et al. Processes in DNA damage response from a whole-cell multi-omics perspective. iScience 2022, 25, 105341. [Google Scholar] [CrossRef]
- Norris, J.L.; Farrow, M.A.; Gutierrez, D.B.; Palmer, L.D.; Muszynski, N.; Sherrod, S.D.; Pino, J.C.; Allen, J.L.; Spraggins, J.M.; Lubbock, A.L.R.; et al. Integrated, high-throughput, multiomics platform enables data-driven construction of cellular responses and reveals global drug mechanisms of action. J. Proteome Res. 2017, 16, 1364–1375. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Neely, M.D.; Davison, C.A.; Aschner, M.; Bowman, A.B. From the Cover: Manganese and Rotenone-Induced Oxidative Stress Signatures Differ in iPSC-Derived Human Dopamine Neurons. Toxicol. Sci. 2017, 159, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.C.; Westlake, G.; Snow, J.P.; Cawthon, B.; Armour, E.; Bowman, A.B.; Ess, K.C. Heterozygous loss of TSC2 alters p53 signaling and human stem cell reprogramming. Hum. Mol. Genet. 2017, 26, 4629–4641. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.H.; Marinelli, N.A.; Shi, Y.; McClatchey, P.M.; Balotin, K.M.; Gullett, D.R.; Hagerla, K.A.; Bowman, A.B.; Ess, K.C.; Wikswo, J.P.; et al. A Simplified, Fully Defined Differentiation Scheme for Producing Blood-Brain Barrier Endothelial Cells from Human iPSCs. Stem Cell Rep. 2019, 12, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef]
- Müller, F.J.; Schuldt, B.M.; Williams, R.; Mason, D.; Altun, G.; Papapetrou, E.P.; Danner, S.; Goldmann, J.E.; Herbst, A.; Schmidt, N.O.; et al. A bioinformatic assay for pluripotency in human cells. Nat. Methods 2011, 8, 315–317. [Google Scholar] [CrossRef]
- Wang, H.; Hao, J.; Hong, C.C. Cardiac induction of embryonic stem cells by a small molecule inhibitor of Wnt/β-catenin signaling. ACS Chem. Biol. 2011, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef]
- Sharma, A.; Li, G.; Rajarajan, K.; Hamaguchi, R.; Burridge, P.W.; Wu, S.M. Derivation of highly purified cardiomyocytes from human induced pluripotent stem cells using small molecule-modulated differentiation and subsequent glucose starvation. J. Vis. Exp. 2015, 97, e52628. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Burridge, P.W.; Kropp, E.M.; Chuppa, S.L.; Kwok, W.M.; Wu, J.C.; Boheler, K.R.; Gundry, R.L. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J. Vis. Exp. 2014, 91, 52010. [Google Scholar] [CrossRef]
- Breckwoldt, K.; Letuffe-Brenière, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Balafkan, N.; Mostafavi, S.; Schubert, M.; Siller, R.; Liang, K.X.; Sullivan, G.; Bindoff, L.A. A method for differentiating human induced pluripotent stem cells toward functional cardiomyocytes in 96-well microplates. Sci. Rep. 2020, 10, 18498. [Google Scholar] [CrossRef] [PubMed]
- Lyra-Leite, D.M.; Gutiérrez-Gutiérrez, Ó.; Wang, M.; Zhou, Y.; Cyganek, L.; Burridge, P.W. A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protoc. 2022, 3, 101560. [Google Scholar] [CrossRef]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179–194.e174. [Google Scholar] [CrossRef]
- Ng, S.Y.; Wong, C.K.; Tsang, S.Y. Differential gene expressions in atrial and ventricular myocytes: Insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. Cell Physiol. 2010, 299, C1234–C1249. [Google Scholar] [CrossRef]
- Ahmann, K.A.; Weinbaum, J.S.; Johnson, S.L.; Tranquillo, R.T. Fibrin degradation enhances vascular smooth muscle cell proliferation and matrix deposition in fibrin-based tissue constructs fabricated in vitro. Tissue Eng. Part A 2010, 16, 3261–3270. [Google Scholar] [CrossRef]
- Schaaf, S.; Shibamiya, A.; Mewe, M.; Eder, A.; Stöhr, A.; Hirt, M.N.; Rau, T.; Zimmermann, W.H.; Conradi, L.; Eschenhagen, T.; et al. Human engineered heart tissue as a versatile tool in basic research and preclinical toxicology. PLoS ONE 2011, 6, e26397. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Eder, A.; Bönstrup, M.; Flato, M.; Mewe, M.; Schaaf, S.; Aksehirlioglu, B.; Schwoerer, A.P.; Uebeler, J.; Eschenhagen, T. Development of a drug screening platform based on engineered heart tissue. Circ. Res. 2010, 107, 35–44. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Maffioletti, S.M.; Sarcar, S.; Henderson, A.B.H.; Mannhardt, I.; Pinton, L.; Moyle, L.A.; Steele-Stallard, H.; Cappellari, O.; Wells, K.E.; Ferrari, G.; et al. Three-Dimensional Human iPSC-Derived Artificial Skeletal Muscles Model Muscular Dystrophies and Enable Multilineage Tissue Engineering. Cell Rep. 2018, 23, 899–908. [Google Scholar] [CrossRef]
- Kerscher, P.; Turnbull, I.C.; Hodge, A.J.; Kim, J.; Seliktar, D.; Easley, C.J.; Costa, K.D.; Lipke, E.A. Direct hydrogel encapsulation of pluripotent stem cells enables ontomimetic differentiation and growth of engineered human heart tissues. Biomaterials 2016, 83, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, P.; Kaczmarek, J.A.; Head, S.E.; Ellis, M.E.; Seeto, W.J.; Kim, J.; Bhattacharya, S.; Suppiramaniam, V.; Lipke, E.A. Direct Production of Human Cardiac Tissues by Pluripotent Stem Cell Encapsulation in Gelatin Methacryloyl. ACS Biomater. Sci. Eng. 2017, 3, 1499–1509. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling expansion and cardiomyogenic differentiation of human pluripotent stem cells in scalable suspension culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes From Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef]
- Halloin, C.; Coffee, M.; Manstein, F.; Zweigerdt, R. Production of Cardiomyocytes from Human Pluripotent Stem Cells by Bioreactor Technologies. Methods Mol. Biol. 2019, 1994, 55–70. [Google Scholar] [CrossRef]
- Freund, C.; Ward-van Oostwaard, D.; Monshouwer-Kloots, J.; van den Brink, S.; van Rooijen, M.; Xu, X.; Zweigerdt, R.; Mummery, C.; Passier, R. Insulin redirects differentiation from cardiogenic mesoderm and endoderm to neuroectoderm in differentiating human embryonic stem cells. Stem Cells 2008, 26, 724–733. [Google Scholar] [CrossRef]
- Lin, Y.; Linask, K.L.; Mallon, B.; Johnson, K.; Klein, M.; Beers, J.; Xie, W.; Du, Y.; Liu, C.; Lai, Y.; et al. Heparin Promotes Cardiac Differentiation of Human Pluripotent Stem Cells in Chemically Defined Albumin-Free Medium, Enabling Consistent Manufacture of Cardiomyocytes. Stem Cells Transl. Med. 2017, 6, 527–538. [Google Scholar] [CrossRef]
- Lin, Y.; Zou, J. Differentiation of Cardiomyocytes from Human Pluripotent Stem Cells in Fully Chemically Defined Conditions. STAR Protoc. 2020, 1, 100015. [Google Scholar] [CrossRef]
- Rosowski, K.A.; Mertz, A.F.; Norcross, S.; Dufresne, E.R.; Horsley, V. Edges of human embryonic stem cell colonies display distinct mechanical properties and differentiation potential. Sci. Rep. 2015, 5, 14218. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Ishioka, C. Inhibition of glycogen synthase kinase-3 beta induces apoptosis and mitotic catastrophe by disrupting centrosome regulation in cancer cells. Sci. Rep. 2015, 5, 13249. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Hosseinabadi, Z.; Salehinejad, P.; Mesbah-Namin, S.A. Differentiation of human adipose-derived stem cells into cardiomyocyte-like cells in fibrin scaffold by a histone deacetylase inhibitor. Biomed. Eng. Online 2017, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.J.; Tatara, A.; Shiu, A.; Sakiyama-Elbert, S.E. Controlled release of neurotrophin-3 and platelet-derived growth factor from fibrin scaffolds containing neural progenitor cells enhances survival and differentiation into neurons in a subacute model of SCI. Cell Transplant. 2010, 19, 89–101. [Google Scholar] [CrossRef]
- Lu, P.; Graham, L.; Wang, Y.; Wu, D.; Tuszynski, M. Promotion of survival and differentiation of neural stem cells with fibrin and growth factor cocktails after severe spinal cord injury. J. Vis. Exp. 2014, 89, e50641. [Google Scholar] [CrossRef]
- Kneser, U.; Voogd, A.; Ohnolz, J.; Buettner, O.; Stangenberg, L.; Zhang, Y.H.; Stark, G.B.; Schaefer, D.J. Fibrin gel-immobilized primary osteoblasts in calcium phosphate bone cement: In vivo evaluation with regard to application as injectable biological bone substitute. Cells Tissues Organs 2005, 179, 158–169. [Google Scholar] [CrossRef]
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: The matrix sandwich method. Circ. Res. 2012, 111, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Möllmann, H.; Nef, H.M.; Kahlert, P.; Kostin, S.; Möllmann, S.; Weber, M.; Troidl, C.; Hamm, C.W.; Holubarsch, C.J.; Elsässer, A. Negative inotropic effect of rapamycin on isolated human cardiomyocytes. J. Int. Med. Res. 2008, 36, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Gonano, L.A.; Jones, P.P. FK506-binding proteins 12 and 12.6 (FKBPs) as regulators of cardiac Ryanodine Receptors: Insights from new functional and structural knowledge. Channels 2017, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Fang, C.X.; Aberle, N.S., 2nd; Ren, B.H.; Ceylan-Isik, A.F.; Ren, J. Inhibition of PI-3 kinase/Akt/mTOR, but not calcineurin signaling, reverses insulin-like growth factor I-induced protection against glucose toxicity in cardiomyocyte contractile function. J. Endocrinol. 2005, 186, 491–503. [Google Scholar] [CrossRef]
- Smyrnias, I.; Mair, W.; Harzheim, D.; Walker, S.A.; Roderick, H.L.; Bootman, M.D. Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell Calcium 2010, 47, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Smyrnias, I.; Thul, R.; Coombes, S.; Roderick, H.L. Atrial cardiomyocyte calcium signalling. Biochim. Biophys. Acta 2011, 1813, 922–934. [Google Scholar] [CrossRef]
- Maxwell, J.T.; Blatter, L.A. A novel mechanism of tandem activation of ryanodine receptors by cytosolic and SR luminal Ca(2+) during excitation-contraction coupling in atrial myocytes. J. Physiol. 2017, 595, 3835–3845. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- MacMillan, D. FK506 binding proteins: Cellular regulators of intracellular Ca2+ signalling. Eur. J. Pharmacol. 2013, 700, 181–193. [Google Scholar] [CrossRef]
- MacMillan, D.; McCarron, J.G. Regulation by FK506 and rapamycin of Ca2+ release from the sarcoplasmic reticulum in vascular smooth muscle: The role of FK506 binding proteins and mTOR. Br. J. Pharmacol. 2009, 158, 1112–1120. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Lamb, R.F. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem. Sci. 2005, 30, 35–42. [Google Scholar] [CrossRef]
- Wang, X.; Yue, P.; Tao, H.; Sun, S.Y. Inhibition of p70S6K does not mimic the enhancement of Akt phosphorylation by rapamycin. Heliyon 2017, 3, e00378. [Google Scholar] [CrossRef] [PubMed]
- Rathmell, J.C.; Fox, C.J.; Plas, D.R.; Hammerman, P.S.; Cinalli, R.M.; Thompson, C.B. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003, 23, 7315–7328. [Google Scholar] [CrossRef]
- Sen, S.; Kundu, B.K.; Wu, H.C.; Hashmi, S.S.; Guthrie, P.; Locke, L.W.; Roy, R.J.; Matherne, G.P.; Berr, S.S.; Terwelp, M.; et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J. Am. Heart Assoc. 2013, 2, e004796. [Google Scholar] [CrossRef] [PubMed]
- Gruppuso, P.A.; Boylan, J.M.; Sanders, J.A. The physiology and pathophysiology of rapamycin resistance: Implications for cancer. Cell Cycle 2011, 10, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Blenis, J. Not all substrates are treated equally: Implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 2009, 8, 567–572. [Google Scholar] [CrossRef]
- Carrière, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef]
- Ito, H.; Ichiyanagi, O.; Naito, S.; Bilim, V.N.; Tomita, Y.; Kato, T.; Nagaoka, A.; Tsuchiya, N. GSK-3 directly regulates phospho-4EBP1 in renal cell carcinoma cell-line: An intrinsic subcellular mechanism for resistance to mTORC1 inhibition. BMC Cancer 2016, 16, 393. [Google Scholar] [CrossRef]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef]
- Harwood, F.C.; Klein Geltink, R.I.; O’Hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Huang, S.; Houghton, P.J. Predicted mechanisms of resistance to mTOR inhibitors. Br. J. Cancer 2006, 95, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Wood, M.A.; Gjertson, C.K.; Katz, H.R.; Burakoff, S.J.; Bierer, B.E. FK506 binding protein 12 mediates sensitivity to both FK506 and rapamycin in murine mast cells. Eur. J. Immunol. 1995, 25, 563–571. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Nellist, M.; Verhaaf, B.; Goedbloed, M.A.; Reuser, A.J.; van den Ouweland, A.M.; Halley, D.J. TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin-hamartin complex. Hum. Mol. Genet. 2001, 10, 2889–2898. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Packard, C.J.; Taskinen, M.R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Dib, I.; Khalil, A.; Chouaib, R.; El-Makhour, Y.; Noureddine, H. Apolipoprotein C-III and cardiovascular diseases: When genetics meet molecular pathologies. Mol. Biol. Rep. 2021, 48, 875–886. [Google Scholar] [CrossRef]
- Zaki, M.E.; Amr, K.S.; Abdel-Hamid, M. APOA2 Polymorphism in Relation to Obesity and Lipid Metabolism. Cholesterol 2013, 2013, 289481. [Google Scholar] [CrossRef] [PubMed]
- Papackova, Z.; Cahova, M. Fatty acid signaling: The new function of intracellular lipases. Int. J. Mol. Sci. 2015, 16, 3831–3855. [Google Scholar] [CrossRef]
- Brown, N.F.; Stefanovic-Racic, M.; Sipula, I.J.; Perdomo, G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism 2007, 56, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X.; Ruiz, J.; White, J.V.; Rossetti, L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Investig. 1994, 93, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Interaction between free fatty acids and glucose metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef]
- Miklas, J.W.; Levy, S.; Hofsteen, P.; Mex, D.I.; Clark, E.; Muster, J.; Robitaille, A.M.; Sivaram, G.; Abell, L.; Goodson, J.M.; et al. Amino acid primed mTOR activity is essential for heart regeneration. iScience 2022, 25, 103574. [Google Scholar] [CrossRef]
- Drake, K.J.; Sidorov, V.Y.; McGuinness, O.P.; Wasserman, D.H.; Wikswo, J.P. Amino acids as metabolic substrates during cardiac ischemia. Exp. Biol. Med. 2012, 237, 1369–1378. [Google Scholar] [CrossRef]
- Wang, C.; Dostanic, S.; Servant, N.; Chalifour, L.E. Egr-1 negatively regulates expression of the sodium-calcium exchanger-1 in cardiomyocytes in vitro and in vivo. Cardiovasc. Res. 2005, 65, 187–194. [Google Scholar] [CrossRef]
- Kasneci, A.; Kemeny-Suss, N.M.; Komarova, S.V.; Chalifour, L.E. Egr-1 negatively regulates calsequestrin expression and calcium dynamics in ventricular cells. Cardiovasc. Res. 2009, 81, 695–702. [Google Scholar] [CrossRef]
- Baron, V.; Adamson, E.D.; Calogero, A.; Ragona, G.; Mercola, D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006, 13, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Medzikovic, L.; Schumacher, C.A.; Verkerk, A.O.; van Deel, E.D.; Wolswinkel, R.; van der Made, I.; Bleeker, N.; Cakici, D.; van den Hoogenhof, M.M.; Meggouh, F.; et al. Orphan nuclear receptor Nur77 affects cardiomyocyte calcium homeostasis and adverse cardiac remodelling. Sci. Rep. 2015, 5, 15404. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Cañes, L.; Martí-Pàmies, I.; Ballester-Servera, C.; Herraiz-Martínez, A.; Alonso, J.; Galán, M.; Nistal, J.F.; Muniesa, P.; Osada, J.; Hove-Madsen, L.; et al. Neuron-derived orphan receptor-1 modulates cardiac gene expression and exacerbates angiotensin II-induced cardiac hypertrophy. Clin. Sci. 2020, 134, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Koren, L.; Elhanani, O.; Kehat, I.; Hai, T.; Aronheim, A. Adult cardiac expression of the activating transcription factor 3, ATF3, promotes ventricular hypertrophy. PLoS ONE 2013, 8, e68396. [Google Scholar] [CrossRef] [PubMed]
- Soraya, A.S.; Tali, H.; Rona, S.; Tom, F.; Roy, K.; Ami, A. ATF3 expression in cardiomyocytes and myofibroblasts following transverse aortic constriction displays distinct phenotypes. Int. J. Cardiol. Heart Vasc. 2021, 32, 100706. [Google Scholar] [CrossRef]
- Huang, L.; Wu, K.H.; Zhang, L.; Wang, Q.; Tang, S.; Wu, Q.; Jiang, P.H.; Lin, J.J.; Guo, J.; Wang, L.; et al. Critical Roles of Xirp Proteins in Cardiac Conduction and Their Rare Variants Identified in Sudden Unexplained Nocturnal Death Syndrome and Brugada Syndrome in Chinese Han Population. J. Am. Heart Assoc. 2018, 7, e006320. [Google Scholar] [CrossRef]
- Mikhailov, A.T.; Torrado, M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int. J. Dev. Biol. 2008, 52, 811–821. [Google Scholar] [CrossRef]
- Piroddi, N.; Pesce, P.; Scellini, B.; Manzini, S.; Ganzetti, G.S.; Badi, I.; Menegollo, M.; Cora, V.; Tiso, S.; Cinquetti, R.; et al. Myocardial overexpression of ANKRD1 causes sinus venosus defects and progressive diastolic dysfunction. Cardiovasc. Res. 2020, 116, 1458–1472. [Google Scholar] [CrossRef]
- Funakoshi, S.; Yoshida, Y. Recent progress of iPSC technology in cardiac diseases. Arch. Toxicol. 2021, 95, 3633–3650. [Google Scholar] [CrossRef]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Kivelä, R. Cardiomyocyte-Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef]
- Sinnecker, D.; Goedel, A.; Laugwitz, K.L.; Moretti, A. Induced pluripotent stem cell-derived cardiomyocytes: A versatile tool for arrhythmia research. Circ. Res. 2013, 112, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on I(K1). Pharmacol. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef]
- Volpato, V.; Webber, C. Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis. Model. Mech. 2020, 13, dmm042317. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorov, V.Y.; Sidorova, T.N.; Samson, P.C.; Reiserer, R.S.; Britt, C.M.; Neely, M.D.; Ess, K.C.; Wikswo, J.P. Contractile and Genetic Characterization of Cardiac Constructs Engineered from Human Induced Pluripotent Stem Cells: Modeling of Tuberous Sclerosis Complex and the Effects of Rapamycin. Bioengineering 2024, 11, 234. https://doi.org/10.3390/bioengineering11030234
Sidorov VY, Sidorova TN, Samson PC, Reiserer RS, Britt CM, Neely MD, Ess KC, Wikswo JP. Contractile and Genetic Characterization of Cardiac Constructs Engineered from Human Induced Pluripotent Stem Cells: Modeling of Tuberous Sclerosis Complex and the Effects of Rapamycin. Bioengineering. 2024; 11(3):234. https://doi.org/10.3390/bioengineering11030234
Chicago/Turabian StyleSidorov, Veniamin Y., Tatiana N. Sidorova, Philip C. Samson, Ronald S. Reiserer, Clayton M. Britt, M. Diana Neely, Kevin C. Ess, and John P. Wikswo. 2024. "Contractile and Genetic Characterization of Cardiac Constructs Engineered from Human Induced Pluripotent Stem Cells: Modeling of Tuberous Sclerosis Complex and the Effects of Rapamycin" Bioengineering 11, no. 3: 234. https://doi.org/10.3390/bioengineering11030234
APA StyleSidorov, V. Y., Sidorova, T. N., Samson, P. C., Reiserer, R. S., Britt, C. M., Neely, M. D., Ess, K. C., & Wikswo, J. P. (2024). Contractile and Genetic Characterization of Cardiac Constructs Engineered from Human Induced Pluripotent Stem Cells: Modeling of Tuberous Sclerosis Complex and the Effects of Rapamycin. Bioengineering, 11(3), 234. https://doi.org/10.3390/bioengineering11030234