

Nanoparticle-Mediated Delivery of Deferasirox: A Promising Strategy Against Invasive Aspergillosis



Abstract

1. Introduction
2. Materials and Methods
2.1. Nanoparticle Formulation
2.2. Particle Size and Zeta Potential Analyses
2.3. Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM)
2.4. Drug Entrapment (DE%) and Drug Loading (DL%) Efficiencies
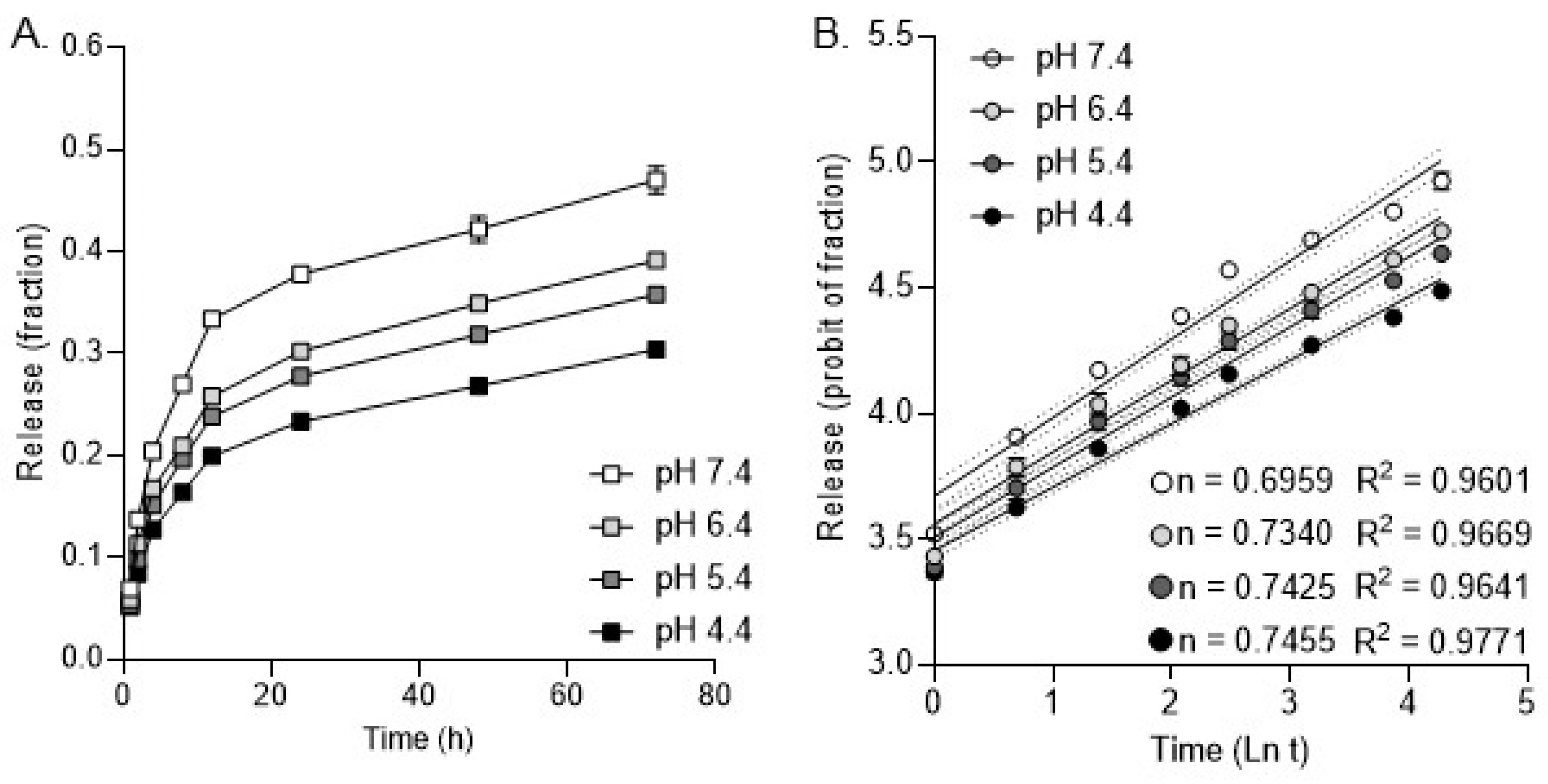
2.5. Release Kinetics Analysis
2.6. Mammalian Cell Culture
2.7. Fungal Culture
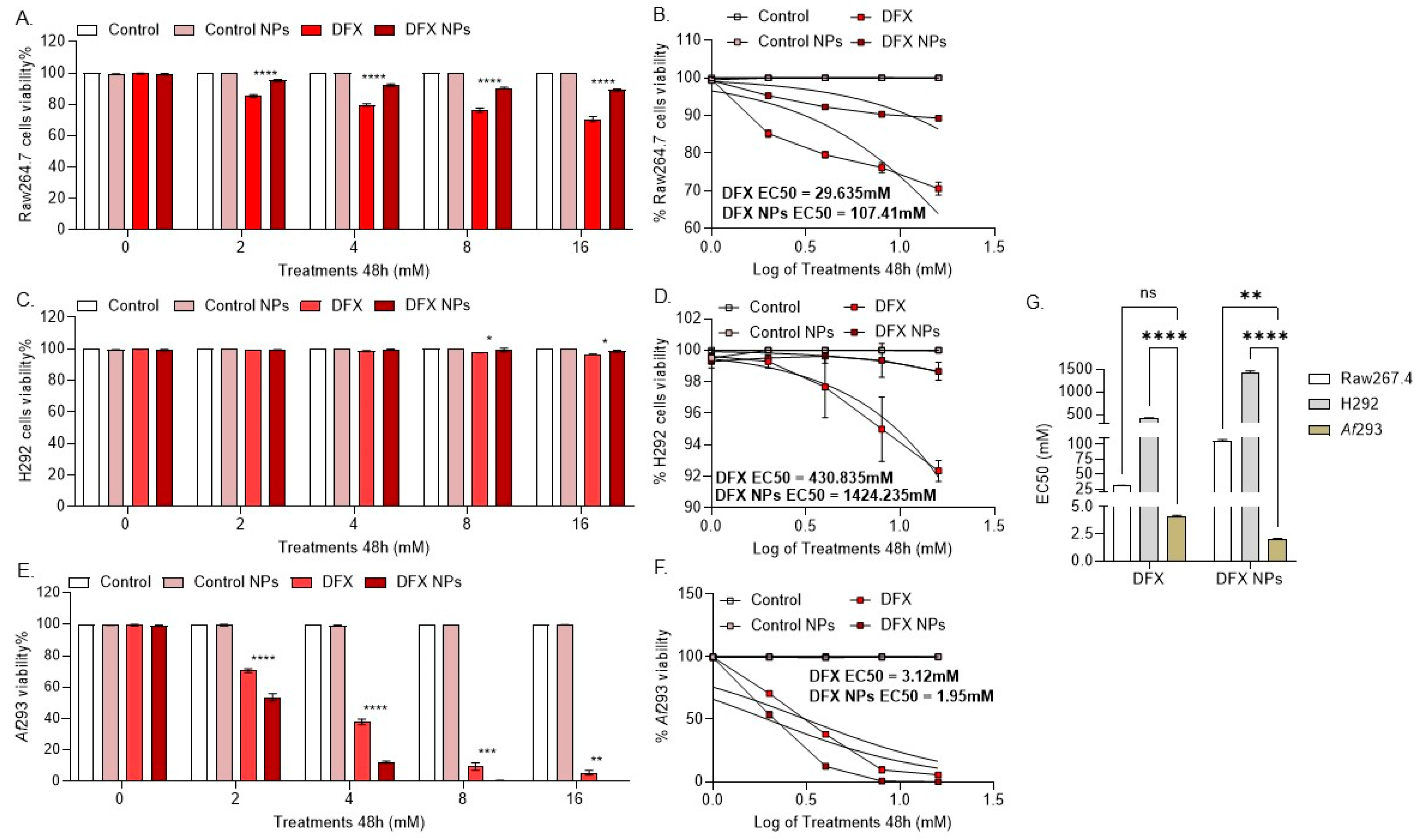
2.8. Cytotoxicity Evaluation
2.8.1. Mammalian Cells
2.8.2. Fungal Cultures
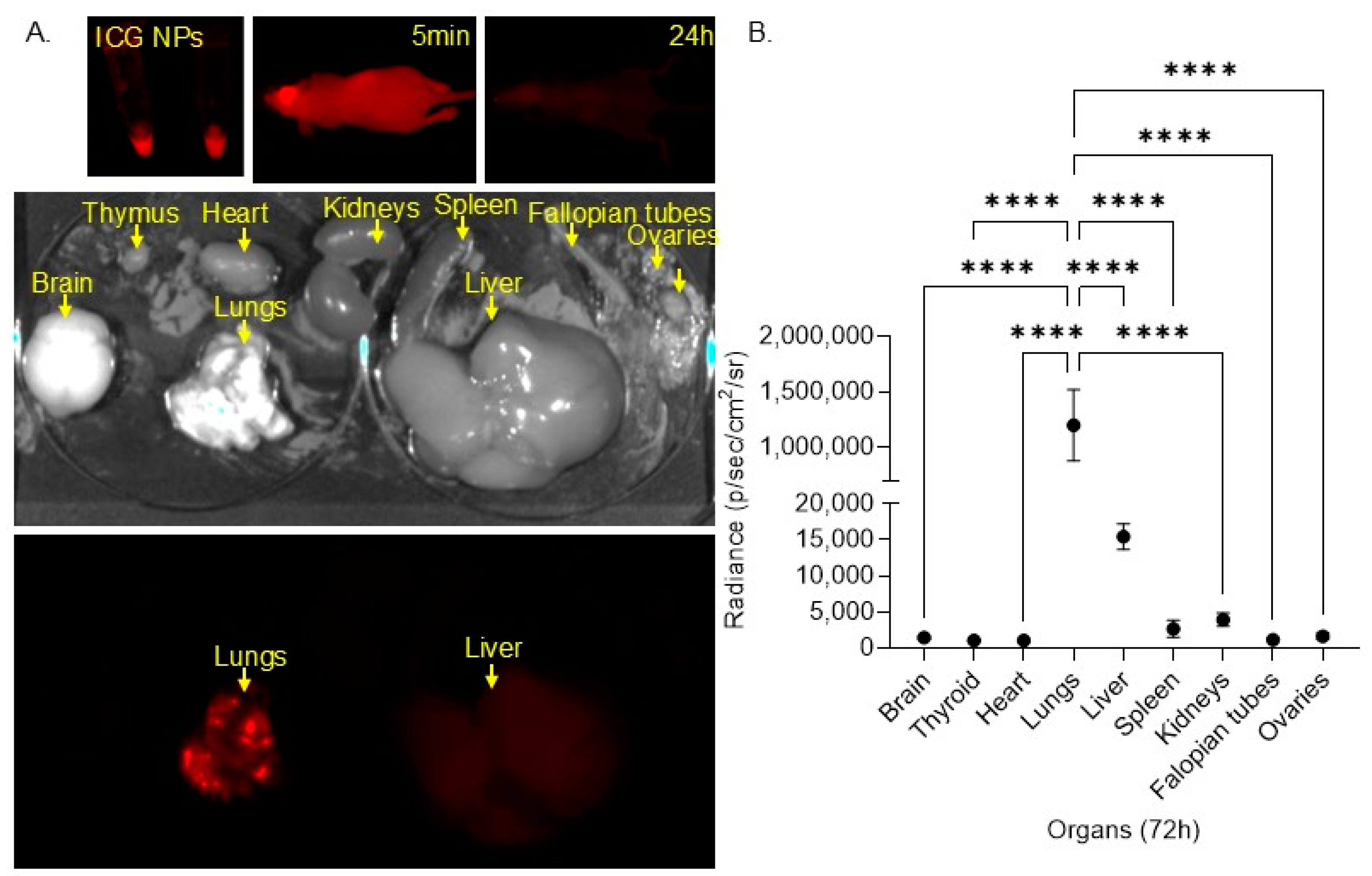
2.9. In Vivo Biodistribution Model
2.10. Neutropenic Mouse Model of IA
2.11. qPCR to Determine Fungal Burden
2.12. Study Approval
2.13. Statistical Analysis
3. Results
Nanoparticle Characterization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takazono, T.; Sheppard, D.C. Aspergillus in chronic lung disease: Modeling what goes on in the airways. In Medical Mycology; Oxford University Press: Oxford, UK, 2017; Volume 55, pp. 39–47. [Google Scholar]
- Cadena, J.; Thompson, G.R.; Patterson, T.F. Aspergillosis: Epidemiology, Diagnosis, and Treatment. Infect. Dis. Clin. N. Am. 2021, 35, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Denning, D.W. Global incidence and mortality of severe fungal disease. Lancet Infect. Dis. 2024, 24, e428–e438. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.H.; Segre, J.A. Cultivating fungal research. Science 2020, 368, 365–366. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Division (AMR); Control of Neglected Tropical Diseases (NTD); Global Coordination and Partnership (GCP). WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Chowdhary, A.; Sharma, C.; Meis, J.F. Azole-resistant aspergillosis: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 2017, 216, S436–S444. [Google Scholar] [CrossRef]
- Burks, C.; Darby, A.; Londoño, L.G.; Momany, M.; Brewer, M.T. Azole-resistant Aspergillus fumigatus in the environment: Identifying key reservoirs and hotspots of antifungal resistance. PLoS Pathog. 2021, 17, e1009711. [Google Scholar] [CrossRef]
- Korfanty, G.; Heifetz, E.; Xu, J. Assessing Thermal Adaptation of a Global Sample of Aspergillus Fumigatus: Implications for Climate Change Effects. Front. Public Health 2023, 11, 1059238. [Google Scholar] [CrossRef]
- Van Rhijn, N.; Bromley, M. The consequences of our changing environment on life threatening and debilitating fungal diseases in humans. J. Fungi 2021, 7, 367. [Google Scholar] [CrossRef]
- Albrich, W.C.; Lamoth, F. Viral-associated Pulmonary Aspergillosis: Have We Finally Overcome the Debate of Colonization versus Infection? Am. J. Respir. Crit. Care Med. 2023, 208, 230–231. [Google Scholar] [CrossRef]
- Marr, K.A.; Platt, A.; Tornheim, J.A.; Zhang, S.X.; Datta, K.; Cardozo, C.; Garcia-Vidal, C. Aspergillosis complicating severe coronavirus disease. Emerg. Infect. Dis. 2021, 27, 18–25. [Google Scholar] [CrossRef]
- Matthaiou, E.I.; Sass, G.; Stevens, D.A.; Hsu, J.L. Iron: An essential nutrient for Aspergillus fumigatus and a fulcrum for pathogenesis. Curr. Opin. Infect. Dis. 2018, 31, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Nazik, H.; Penner, J.C.; Ferreira, J.A.; Haagensen, J.A.J.; Cohen, K.; Spormann, A.M.; Martinez, M.; Chen, V.; Hsu, J.L.; Clemons, K.V.; et al. Effects of iron chelators on the formation and development of aspergillus fumigatus biofilm. Antimicrob. Agents Chemother. 2015, 59, 6514–6520. [Google Scholar] [CrossRef] [PubMed]
- Haas, H. Iron—A key nexus in the virulence of Aspergillus fumigatus. Front. Microbiol. 2012, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, G.; Jung, W.H.; Kronstad, J.W. Iron acquisition in fungal pathogens of humans. Metallomics 2017, 9, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Corrales, J.; Ramos-Alonso, L.; González-Sabín, J.; Ríos-Lombardía, N.; Trevijano-Contador, N.; Berg, H.E.; Skottvoll, F.S.; Moris, F.; Zaragoza, O.; Chymkowitch, P.; et al. Characterization of a selective, iron-chelating antifungal compound that disrupts fungal metabolism and synergizes with fluconazole. Microbiol. Spectr. 2024, 12, e0259423. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Spellberg, B.; Edwards, J.J. Iron acquisition: A novel perspective on mucormycosis pathogenesis and treatment. Curr. Opin. Infect. Dis. 2008, 21, 620–625. [Google Scholar] [CrossRef]
- Schwarz, P.; Cornely, O.A.; Dannaoui, E. Antifungal combinations in Mucorales: A microbiological perspective. Mycoses 2019, 62, 746–760. [Google Scholar] [CrossRef]
- Zarember, K.A.; Cruz, A.R.; Huang, C.-Y.; Gallin, J.I. Antifungal activities of natural and synthetic iron chelators alone and in combination with azole and polyene antibiotics against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2009, 53, 2654–2656. [Google Scholar] [CrossRef]
- iMagIng pulmonaRy Aspergillosis Using Gallium-68-dEferoxamine (MIRAGE). Available online: https://ctv.veeva.com/study/imaging-pulmonary-aspergillosis-using-gallium-68-deferoxamine (accessed on 30 September 2024).
- Fang, W.; Wu, J.; Cheng, M.; Zhu, X.; Du, M.; Chen, C.; Liao, W.; Zhi, K.; Pan, W. Diagnosis of invasive fungal infections: Challenges and recent developments. J. Biomed. Sci. 2023, 30, 42. [Google Scholar] [CrossRef]
- Sun, J.; Xiao, S.; Xue, C. The tug-of-war on iron between plant and pathogen. Phytopathol. Res. 2023, 5, 61. [Google Scholar] [CrossRef]
- Miethke, M. Molecular strategies of microbial iron assimilation: From high-affinity complexes to cofactor assembly systems. Metallomics 2013, 5, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Lunawat, A.K.; Kumar, A.; Sharma, T.; Islam, M.; Kahlon, M.S.; Mukherjee, D.; Narang, R.K.; Raikwar, S. Recent Trends in Nanocarrier-Based Drug Delivery System for Prostate Cancer. AAPS PharmSciTech 2024, 25, 55. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, L.; Fu, Y. Nanotechnology-based ocular drug delivery systems: Recent advances and future prospects. J. Nanobiotechnology 2023, 21, 232. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudnia, N.; Eftekhari, R.B.; Sohi, A.N.; Zamzami, A.; Dorkoosh, F.A. Application of nano-based systems for drug delivery and targeting: A review. J. Nanoparticle Res. 2020, 22, 245. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Forest, V.; Pourchez, J. Human biological monitoring of nanoparticles, a new way to investigate potential causal links between exposure to nanoparticles and lung diseases? Pulmonology 2022, 29, 4–5. [Google Scholar] [CrossRef]
- Zou, W.; Liu, C.; Chen, Z.; Zhang, N. Studies on bioadhesive PLGA nanoparticles: A promising gene delivery system for efficient gene therapy to lung cancer. Int. J. Pharm. 2009, 370, 187–195. [Google Scholar] [CrossRef]
- Musumeci, T.; Ventura, C.; Giannone, I.; Ruozi, B.; Montenegro, L.; Pignatello, R.; Puglisi, G. PLA/PLGA nanoparticles for sustained release of docetaxel. Int. J. Pharm. 2006, 325, 172–179. [Google Scholar] [CrossRef]
- Yang, J.; Zeng, H.; Luo, Y.; Chen, Y.; Wang, M.; Wu, C.; Hu, P. Recent Applications of PLGA in Drug Delivery Systems. Polymers 2024, 16, 2606. [Google Scholar] [CrossRef]
- Chaudhary, A.; Shambhakar, S. Nanotechnology in Drug Delivery: Overcoming Poor Solubility Challenges through Nanoformulations. Curr. Nanomed. 2024, 14, 200–211. [Google Scholar] [CrossRef]
- Lu, Y.; Cheng, D.; Niu, B.; Wang, X.; Wu, X.; Wang, A. Properties of Poly (Lactic-co-Glycolic Acid) and Progress of Poly (Lactic-co-Glycolic Acid)-Based Biodegradable Materials in Biomedical Research. Pharmaceuticals 2023, 16, 454. [Google Scholar] [CrossRef] [PubMed]
- Rahdar, A.; Hajinezhad, M.R.; Sargazi, S.; Barani, M.; Bilal, M.; Kyzas, G.Z. Deferasirox-loaded pluronic nanomicelles: Synthesis, characterization, in vitro and in vivo studies. J. Mol. Liq. 2020, 323, 114605. [Google Scholar] [CrossRef]
- Rao Somisetty, V.S.; Kumar Bichala, P.; Rao, C.P.; Kirankumar, V. Development and validation of newer analytical methods for the estimation of deferasirox in bulk and in tablet dosage form by calorimetric method. Int. J. Pharm. Pharm. Sci. 2013, 5, 521–525. [Google Scholar]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Perez, R.P.; Godwin, A.K.; Handel, L.M.; Hamilton, T.C. A Comparison of Clonogenic, Microtetrazolium and Sulforhodamine B Assays for Determination of Cisplatin Cytotoxicity in Human Ovarian Carcinoma Cell Lines. Eur. J. Cancer 1993, 29, 395–399. [Google Scholar] [CrossRef]
- Meletiadis, J.; Mouton, J.W.; Meis, J.F.G.M.; Bouman, B.A.; Donnelly, J.P.; Verweij, P.E.; Network, E. Colorimetric assay for antifungal susceptibility testing of Aspergillus Species. J. Clin. Microbiol. 2001, 39, 3402–3408. [Google Scholar] [CrossRef]
- Loures, F.V.; Levitz, S.M. XTT Assay of Antifungal Activity Materials and Reagents. Bio-protocol 2015, 5, e1543. [Google Scholar] [CrossRef]
- Sass, G.; Kotta-Loizou, I.; Martinez, M.; Larwood, D.J.; Stevens, D.A. Polymycovirus Infection Sensitizes Aspergillus fumigatus for Antifungal Effects of Nikkomycin Z. Viruses 2023, 15, 197. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Cho, Y.-W.; Yoon, J.; Song, S.-G.; Noh, Y.-W. Mitochondrial DNA as a target for analyzing the biodistribution of cell therapy products. Sci. Rep. 2024, 14, 7934. [Google Scholar] [CrossRef]
- Giddabasappa, A.; Gupta, V.R.; Norberg, R.; Gupta, P.; Spilker, M.E.; Wentland, J.; Rago, B.; Eswaraka, J.; Leal, M.; Sapra, P. Biodistribution and targeting of anti-5t4 antibody–drug conjugate using fluorescence molecular tomography. Mol. Cancer Ther. 2016, 15, 2530–2540. [Google Scholar] [CrossRef]
- Vasquez, K.O.; Casavant, C.; Peterson, J.D. Quantitative whole body biodistribution of fluorescent-labeled agents by non-invasive tomographic imaging. PLoS ONE 2011, 6, e20594. [Google Scholar] [CrossRef]
- Tansi, F.L.; Rüger, R.; Kollmeier, A.M.; Böhm, C.; Kontermann, R.E.; Teichgraeber, U.K.; Fahr, A.; Hilger, I. A fast and effective determination of the biodistribution and subcellular localization of fluorescent immunoliposomes in freshly excised animal organs. BMC Biotechnol. 2017, 17, 8. [Google Scholar] [CrossRef]
- Lote, C.J. Principles of Renal Physiology; Chapman & Hall: London, UK, 1994. [Google Scholar]
- Rippke, F.; Berardesca, E.; Weber, T.M. 87–94: pH and Microbial Infections. In pH of the Skin: Issues and Challenges; S. Karger AG: Basel, Switzerland, 2018. [Google Scholar]
- Wang, J.; MacEwan, S.R.; Chilkoti, A. Quantitative Mapping of the Spatial Distribution of Nanoparticles in Endo-Lysosomes by Local pH. Nano Lett. 2017, 17, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Seifter, J.L. Body Fluid Compartments, Cell Membrane Ion Transport, Electrolyte Concentrations, and Acid-Base Balance. Semin. Nephrol. 2019, 39, 368–379. [Google Scholar] [CrossRef]
- Beasley, D.E.; Koltz, A.M.; Lambert, J.E.; Fierer, N.; Dunn, R.R. The evolution of stomach acidity and its relevance to the human microbiome. PLoS ONE 2015, 10, e0134116. [Google Scholar] [CrossRef] [PubMed]
- Metzetti, M.; Soloperto, M.; Fasoli, A.; Mattoli, S.; Italy, M. Presented in Abstract Form. In Proceedings of the World Health Conference on Lung Health, Boston, MA, USA, 20–24 May 1990. [Google Scholar]
- Gow, N.A.R.; Latge, J.-P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 2017, 5, 28513415. [Google Scholar] [CrossRef]
- Beauvais, A.; Fontaine, T.; Aimanianda, V.; Latgé, J.-P. Aspergillus cell wall and biofilm. Mycopathologia 2014, 178, 371–377. [Google Scholar] [CrossRef]
- Cong, X.; Zhang, Z.; Li, H.; Yang, Y.-G.; Zhang, Y.; Sun, T. Nanocarriers for targeted drug delivery in the vascular system: Focus on endothelium. J. Nanobiotechnol. 2024, 22, 1–29. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.; Li, J.; Nie, Y.; Liao, G.; Yu, Y.; Li, C. Overcoming the Reticuloendothelial System Barrier to Drug Delivery with a “Don’t-Eat-Us” Strategy. ACS Nano 2019, 13, 13015–13026. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-K.; Utsumi, T.; Seo, Y.-E.; Deng, Y.; Satoh, A.; Saltzman, W.M.; Iwakiri, Y. Cellular distribution of injected PLGA-nanoparticles in the liver. Nanomed. Nanotechnology Biol. Med. 2016, 12, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yang, S.-G.; Shim, W.-S.; Cui, F.; Cheng, G.; Kim, I.-W.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Lung-Specific Delivery of Paclitaxel by Chitosan-Modified PLGA Nanoparticles Via Transient Formation of Microaggregates. J. Pharm. Sci. 2009, 98, 970–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W. Nanoparticle Aggregation: Principles and Modeling. In Nanomaterial: Impacts on Cell Biology and Medicine; Capco, D.G., Chen, Y., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 19–43. [Google Scholar] [CrossRef]
- Praphawatvet, T.; Williams, R.O. Precipitation Technologies for Nanoparticle Production. In Formulating Poorly Water Soluble Drugs; Williams, R.O., III, Davis, D.A., Jr., Miller, D.A., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 529–598. [Google Scholar] [CrossRef]
- Guérin, M.; Lepeltier, E. Nanomedicines via the pulmonary route: A promising strategy to reach the target? Drug Deliv. Transl. Res. 2024, 14, 2276–2297. [Google Scholar] [CrossRef]
- Lu, X.; Zhu, T.; Chen, C.; Liu, Y. Right or Left: The role of nanoparticles in pulmonary diseases. Int. J. Mol. Sci. 2014, 15, 17577–17600. [Google Scholar] [CrossRef] [PubMed]
- Stauber, H.; Waisman, D.; Korin, N.; Sznitman, J. Red blood cell dynamics in biomimetic microfluidic networks of pulmonary alveolar capillaries. Biomicrofluidics 2017, 11, 014103. [Google Scholar] [CrossRef]
- Huang, Y.; Doerschuk, C.M.; Kamm, R.D.; Tawhai, M.H.; Bates, J.H.T.; Clark, A.R.; Hoffman, E.A.; Burrowes, K.S.; Zhao, M.; Fernandez, L.G.; et al. Computational Modeling of RBC and Neutrophil Transit through the Pulmonary Capillaries. J. Appl. Physiol. 2001, 90, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Murata, J.; Unekawa, M.; Kudo, Y.; Kotani, M.; Kanno, I.; Izawa, Y.; Tomita, Y.; Tanaka, K.F.; Nakahara, J.; Masamoto, K. Acceleration of the Development of Microcirculation Embolism in the Brain due to Capillary Narrowing. Stroke 2023, 54, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Fornaguera, C.; Calderó, G.; Mitjans, M.; Vinardell, M.P.; Solans, C.; Vauthier, C. Interactions of PLGA nanoparticles with blood components: Protein adsorption, coagulation, activation of the complement system and hemolysis studies. Nanoscale 2015, 7, 6045–6058. [Google Scholar] [CrossRef]
- Hassan, S.A. Computational Study of the Forces Driving Aggregation of Ultrasmall Nanoparticles in Biological Fluids. ACS Nano 2017, 11, 4145–4154. [Google Scholar] [CrossRef]
- Alvi, M.; Yaqoob, A.; Rehman, K.; Shoaib, S.M.; Akash, M.S.H. PLGA-based nanoparticles for the treatment of cancer: Current strategies and perspectives. AAPS Open 2022, 8, 12. [Google Scholar] [CrossRef]
- Zhou, J.; Li, D.; Wen, H.; Zheng, S.; Su, C.; Yi, F.; Wang, J.; Liang, Z.; Tang, T.; Zhou, D.; et al. Inter-molecular β-sheet structure facilitates lung-targeting siRNA delivery. Sci. Rep. 2016, 6, 22731. [Google Scholar] [CrossRef] [PubMed]
- Rivera, K.A.; Gao, D.; Holloway, J.; Li, S.; Johnson, S.R.; Szekely, Z.; Sinko, P.J. Passive Pulmonary Targeting: Preliminary Assessment of the Effects of Microparticle Deposition in the Lungs. FASEB J. 2016, 30, 933.3. [Google Scholar] [CrossRef]
- Garanina, A.; Vishnevskiy, D.; Chernysheva, A.; Malinovskaya, J.; Lazareva, P.; Semkina, A.; Abakumov, M.; Naumenko, V. The Internalization Pathways of Liposomes, PLGA, and Magnetic Nanoparticles in Neutrophils. Biomedicines 2024, 12, 2180. [Google Scholar] [CrossRef] [PubMed]
- Kutscher, H.L.; Chao, P.; Deshmukh, M.; Singh, Y.; Hu, P.; Joseph, L.B.; Reimer, D.C.; Stein, S.; Laskin, D.L.; Sinko, P.J. Threshold size for optimal passive pulmonary targeting and retention of rigid microparticles in rats. J. Control. Release 2010, 143, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, S.; Zhao, G.; Fu, X.; Xie, X.; Huang, Y.; Cheng, X.; Wei, J.; Liu, H.; Lai, Z. Long-term intravenous administration of carboxylated single-walled carbon nanotubes induces persistent accumulation in the lungs and pulmonary fibrosis via the nuclear factor-kappa b pathway. Int. J. Nanomed. 2017, 12, 263–277. [Google Scholar] [CrossRef]
- Kreyling, W.G.; Holzwarth, U.; Haberl, N.; Kozempel, J.; Wenk, A.; Hirn, S.; Schleh, C.; Schäffler, M.; Lipka, J.; Semmler-Behnke, M.; et al. Quantitative biokinetics of titanium dioxide nanoparticles after intratracheal instillation in rats: Part 3. Nanotoxicology 2017, 11, 454–464. [Google Scholar] [CrossRef]
- Emami, F.; Yazdi, S.J.M.; Na, D.H. Poly(lactic acid)/poly(lactic-co-glycolic acid) particulate carriers for pulmonary drug delivery. J. Pharm. Investig. 2019, 49, 427–442. [Google Scholar] [CrossRef]
- Geiser, M.; Wigge, C.; Conrad, M.L.; Eigeldinger-Berthou, S.; Künzi, L.; Garn, H.; Renz, H.; A Mall, M. Nanoparticle uptake by airway phagocytes after fungal spore challenge in murine allergic asthma and chronic bronchitis. BMC Pulm. Med. 2014, 14, 116. [Google Scholar] [CrossRef]
- Geiser, M. Morphological aspects of particle uptake by lung phagocytes. Microsc. Res. Tech. 2002, 57, 512–522. [Google Scholar] [CrossRef]
- Geiser, M. Update on Macrophage Clearance of Inhaled Micro- and Nanoparticles. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 207–217. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Geiser, M.; Gerber, P.; Maye, I.; Hof, V.I.; Gehr, P. Retention of Teflon Particles in Hamster Lungs: A Stereological Study. J. Aerosol Med. 2000, 13, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Nikula, K.J.; Avila, K.J.; Griffith, W.C.; Mauderly, J.L. Sites of Particle Retention and Lung Tissue Responses to Chronically Inhaled Diesel Exhaust and Coal Dust in Rats and Cynomolgus Monkeys. Environ. Health Perspect. 1997, 105, 1231–1234. [Google Scholar] [PubMed]
- Yin, D.; Zhang, M.; Chen, J.; Huang, Y.; Liang, D. Shear-responsive peptide/siRNA complexes as lung-targeting gene vectors. Chin. Chem. Lett. 2020, 32, 1731–1736. [Google Scholar] [CrossRef]
- Brauer, M.; Avila-Casado, C.; Fortoul, T.I.; Vedal, S.; Stevens, B.; Churg, A. Air Pollution and Retained Particles in the Lung. Environ. Health Perspect. 2001, 109, 1039–1043. Available online: http://ehpnet1.niehs.nih.gov/docs/2001/109p1039-1043brauer/abstract.html (accessed on 28 October 2024). [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Equation | DFX NPs |
|---|---|
| DL% = ((Dt mass − Df mass)/NP mass) × 100 | 77.9% |
| DE% = ((Dt mass − Df mass)/Dt mass) × 100 | 79.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peppe, S.; Farrokhi, M.; Waite, E.A.; Muhi, M.; Matthaiou, E.I. Nanoparticle-Mediated Delivery of Deferasirox: A Promising Strategy Against Invasive Aspergillosis. Bioengineering 2024, 11, 1115. https://doi.org/10.3390/bioengineering11111115
Peppe S, Farrokhi M, Waite EA, Muhi M, Matthaiou EI. Nanoparticle-Mediated Delivery of Deferasirox: A Promising Strategy Against Invasive Aspergillosis. Bioengineering. 2024; 11(11):1115. https://doi.org/10.3390/bioengineering11111115
Chicago/Turabian StylePeppe, Sydney, Moloud Farrokhi, Evan A. Waite, Mustafa Muhi, and Efthymia Iliana Matthaiou. 2024. "Nanoparticle-Mediated Delivery of Deferasirox: A Promising Strategy Against Invasive Aspergillosis" Bioengineering 11, no. 11: 1115. https://doi.org/10.3390/bioengineering11111115
APA StylePeppe, S., Farrokhi, M., Waite, E. A., Muhi, M., & Matthaiou, E. I. (2024). Nanoparticle-Mediated Delivery of Deferasirox: A Promising Strategy Against Invasive Aspergillosis. Bioengineering, 11(11), 1115. https://doi.org/10.3390/bioengineering11111115