1. Introduction
Among the fundamental reactions critical to contemporary hydrogen production technologies, the water–gas shift (WGS) reaction plays a pivotal role, not only as a key step in enhancing hydrogen yield but also as a strategic method for reducing carbon monoxide concentrations within synthesis gas streams. The water–gas shift reaction is represented by the equation.
This reaction is significant across various industrial applications. In hydrogen production, the WGS reaction is indispensable in reforming processes where hydrogen is extracted from hydrocarbons. In ammonia production, it plays a crucial role in reducing CO concentrations in synthesis gas, which is a vital step in ammonia synthesis. Furthermore, the hydrogen produced through this reaction serves as a clean energy source in fuel cells [
1,
2]. This process is commonly facilitated by redox-active oxides, such as ceria (CeO
2), which play a crucial role in significantly enhancing the reaction efficiency. The presence of these oxides, owing to their redox properties, promotes key reaction steps, thereby improving both the overall reaction rate and selectivity. Despite these advantages, CeO
2-based materials may exhibit certain limitations when applied under realistic operational environments. From a materials chemistry and energy application perspective, issues such as phase instability under high-temperature or reductive conditions, vulnerability to surface contamination, and the necessity for lattice or morphological optimization remain critical challenges. Moreover, the propensity for side reactions, along with the relatively high synthesis and processing costs of nanostructured CeO
2, may further restrict its large-scale deployment in catalytic and energy conversion systems. Therefore, the development of modified or composite oxide systems, designed to retain the redox benefits of ceria while overcoming its intrinsic limitations, has become a focal point of current research [
3,
4,
5,
6,
7]. A promising strategy to enhance CeO
2 performance involves doping with other cations to create additional oxygen vacancies and improve resistance to thermal sintering. For example, a study conducted in 2024 demonstrated that the modification of CeO
2 with Ni can alter its structural and chemical properties by increasing the concentration of oxygen vacancies, thereby enhancing its catalytic potential under demanding operating conditions [
8]. Similarly, a 2023 study revealed that doping CeO
2 with rare earth elements can significantly improve its oxygen storage capacity, leading to enhanced performance in various catalytic and energy-related applications [
9]. Among these rare earth elements, praseodymium (Pr) is particularly noteworthy due to its unique electronic configuration and facile valence fluctuation between Pr
3+ and Pr
4+. This property allows Pr to effectively promote the formation of highly mobile oxygen vacancies within the ceria lattice, significantly boosting the oxygen storage capacity (OSC) and oxygen mobility crucial for the redox cycle mechanism of the WGS reaction. Studies have shown that Pr-doped CeO
2 exhibits superior redox properties and catalytic activity compared to undoped CeO
2 due to the synergistic effect of Pr’s variable oxidation states and the induced lattice defects [
10,
11]. Furthermore, the incorporation of Pr has been demonstrated to enhance the thermal stability of CeO
2-based catalysts, effectively inhibiting particle growth and sintering at elevated temperatures, which is a common challenge impacting long-term catalytic performance [
12]. In addition, noble metals such as Pd, Pt, Rh, and Au are extensively employed in the water–gas shift (WGS) reaction, owing to their superior catalytic activity and selectivity. These metals are frequently incorporated into CeO
2-based systems to further enhance catalytic efficiency. Among various transition metals investigated, cobalt (Co) has garnered significant attention due to its relatively low cost and outstanding catalytic performance in energy-related reactions. However, cobalt catalysts often face challenges, including thermal sintering at high temperatures and susceptibility to oxidation, which can degrade their catalytic performance over time [
13]. To address these issues, the incorporation of additional stabilizing elements, such as rhenium (Re), has been explored to enhance the stability and resistance to sintering of Co-based catalysts [
14,
15].
This study focuses on the improvement of the WGS performance of Co catalysts, utilizing Co in conjunction with rhenium (Re) on CeO
2-based materials as a bimetallic catalyst. The selection of rhenium is based on its unique synergistic properties with both Co and CeO
2 that directly address the inherent limitations of Co catalysts. Specifically, Re has garnered significant attention in recent research due to its ability to form strong interactions with CeO
2, often leading to a “strong metal–support interaction” (SMSI)-like effect, where ReOx species can anchor and stabilize cobalt nanoparticles. This interaction facilitates the uniform dispersion of cobalt, preventing agglomeration and maintaining Co as small, highly active nanoparticles, thereby significantly mitigating thermal sintering at elevated temperatures [
16,
17,
18].
Furthermore, rhenium plays a crucial role in enhancing the reducibility of cobalt oxides, often facilitating the reduction of Co
3+ to active Co
2+ or metallic Co
0 at lower temperatures, which is essential for catalytic activity and also contributes to improved resistance against re-oxidation during reaction [
19,
20]. Additionally, the incorporation of Re has been shown to enhance the performance of CeO
2 in promoting carbon oxidation by utilizing the oxygen stored in its lattice. This increased capacity for oxygen transfer and consumption effectively reduces the propensity for carbonaceous (coke) deposition on the cobalt surface, which is a major cause of catalyst deactivation in WGS reactions [
21]. The synergistic electronic modifications induced by Re doping also influence the Co-CeO
2 interface, optimizing the adsorption and activation of reactants and further contributing to enhanced catalytic efficiency and stability. Co and Co-Re catalysts supported on CeO
2-based materials were synthesized using impregnation methods, aiming to optimize their structural and catalytic properties for water–gas shift reaction to enhance hydrogen production. In addition, the physical and chemical properties of the synthesized catalysts were characterized using various analytical techniques, including X-ray powder diffraction (XRD), Brunauer–Emmett–Teller (BET) surface area, Scanning Electron Microscopy (SEM), H
2-temperature programmed reduction (H
2-TPR), NH
3-temperature programmed desorption (NH
3-TPD), and X-ray photoelectron spectroscopy (XPS). XRD is utilized to identify the crystalline phases, while BET analysis provides crucial surface area measurements that influence catalytic activity. SEM provides detailed information about the surface morphology and particle size of the catalysts. TPR evaluates the reducibility of the catalysts, and NH
3-TPD assesses CO adsorption capacity. These results were key factors for determining catalyst performance in the WGS reaction.
2. Experimental
2.1. Materials
Cerium(III) nitrate hexahydrate (Ce(NO3)3•6H2O, 99.5% purity, Alfa Aesar, Ward Hill, MA, USA), samarium(III) nitrate hexahydrate (Sm(NO3)3•6H2O, 99.9% purity, Aldrich, Milwaukee, WI, USA), praseodymium(III) nitrate hexahydrate (Pr(NO3)3•6H2O, 99.9% purity, Aldrich), gadolinium(III) nitrate hexahydrate (Gd(NO3)3•6H2O, 99.9%, Alfa Aesar), urea (NH2CONH2, 99% purity, Aldrich), cobalt(II) nitrate hexahydrate (Co(NO3)2•6H2O, 98% purity, Ajax Finechem, Taren Point, New South Wales, Australia), and ammonium perrhenate (NH4ReO4, 99% purity, Aldrich).
2.2. Catalyst Synthesis
CeO2, Ce-5%Sm-O, Ce-5%Gd-O, and Ce-5%Pr-O supports were synthesized using the combustion method. Ce(NO3)3•6H2O and M(NO3)3•6H2O (M = Sm, Gd and Pr) were used as metal precursors, with NH2CONH2 serving as the fuel for the auto-ignition process. The concentration of metal (M) mixed with the cerium oxide support was maintained at 5 wt%. The precursor solution was prepared by dissolving the nitrate salts in a minimal volume of deionized water, followed by heating over a Bunsen burner until auto-ignition occurred.
Monometallic and bimetallic catalysts were prepared using impregnation method. Monometallic Co catalysts were synthesized from cobalt(II) nitrate hexahydrate (Co(NO3)2•6H2O, 98% purity, Ajax Finechem), corresponding to 5 wt.% Co loading. The precursor was dissolved in a minimal volume of deionized water and then impregnated onto CeO2 or doped-CeO2 supports. All catalysts were dried at 110 °C for 12 h, followed by calcination at 650 °C for 8 h.
Bimetallic CoRe catalysts were prepared from Co(NO3)2•6H2O and ammonium perrhenate (NH4ReO4), achieving Co and Re loadings of 4 wt.% and 1 wt.%, respectively. These precursors were dissolved in a minimal volume of deionized water and impregnated onto CeO2 and doped-CeO2 supports. The drying and calcination conditions for the bimetallic catalysts were identical to those used for the monometallic catalysts.
2.3. Catalyst Characterization
X-ray powder diffraction (XRD) is a powerful analytical technique used to determine the crystallographic structure of materials. In XRD, Cu Kα radiation is employed with a wavelength of 1.5406 Å. The operating conditions include an applied voltage of 40 kV and a current of 40 mA and a 2θ range of 20 to 90 degrees, with a step size of 0.02 degrees and a time per step of 0.5 s. The resulting diffraction patterns are analyzed to identify the phase compositions and crystallite sizes of the studied material.
Brunauer–Emmett–Teller (BET) surface area and average pore size measurements are essential techniques for characterizing porous materials. These measurements were conducted using nitrogen adsorption–desorption isotherms at 77.3 K using a BELSORP MAX instrument (MicrotracBEL, Japan). Prior to measurement, the catalysts were degassed under a vacuum at 300 °C for 3 h. The BET method was applied to calculate the specific surface area of the solid materials, while the pore size distribution was derived from the desorption branch of the isotherm data using the Barrett–Joyner–Halenda (BJH) method.
A Scanning Electron Microscope (SEM, Thermo Scientific Phenom ProX G6, Eindhoven, The Netherlands), equipped with an Energy Dispersive X-ray Spectroscopy (EDS) detector, was used to examine the morphology and elemental distribution of the as-synthesized catalysts. Prior to analysis, catalyst samples were prepared by sprinkling the powder onto conductive carbon tape affixed to aluminum stubs. The samples were subsequently coated with a thin layer of gold to ensure conductivity. SEM images were acquired at various magnifications to observe the surface morphology and overall particle distribution. Elemental mapping was performed using EDS to determine the spatial distribution and relative abundance of constituent elements (e.g., Co, Ce, Pr, Re, O) across the catalyst surface.
The H2 temperature-programmed reduction (H2-TPR) measurements were conducted using a BELCAT-B instrument (MicrotracBEL, Osaka, Japan), with 50 mg of the catalyst sample placed in a quartz reactor. The catalyst underwent a pretreatment with a helium (He) flow at 140 °C for 45 min. After pretreatment, the catalyst was cooled to 45 °C in preparation for the reduction phase. Reduction proceeded using hydrogen (H2) in an argon carrier gas at a flow rate of 40 mL/min, with the temperature ramping from 45 °C to 950 °C at 10 °C/min. The H2-TPR profile obtained during the experiment provides valuable information on the reducibility of metal species in the catalyst and the temperature at which reduction occurs.
Raman spectrometry analysis of the Co catalysts was carried out by Perkin Elmer System 2000 FT-IR/FT-Raman. The spectra were collected using argon ion laser irradiation in the range of 100–1500 cm−1 with an output power of 10 mW and wavelength of 532 nm.
NH3 temperature-programmed desorption (NH3-TPD) analysis was performed using a BELCAT-B instrument (MicrotracBEL, Japan) to investigate the strength and distribution of acidic sites on the catalyst surface. In each experiment, 50 mg of the reduced catalyst was pretreated under a helium flow (50 mL/min) at 450 °C for 65 min to remove surface impurities and activate the catalyst. After cooling to 50 °C, an NH3/He gas mixture was introduced for 60 min to allow ammonia adsorption. The TPD profile was recorded by heating the sample from 50 °C to 400 °C at a rate of 10 °C/min under a continuous helium flow of 50 mL/min. The desorbed NH3 was monitored using a thermal conductivity detector (TCD).
The X-ray photoelectron spectroscopy (XPS) experiment examines the core-level oxygen (O 1s) electrons in a material to determine its composition. This technique is valuable for surface analysis, providing insights into the presence of different oxygen-containing species.
2.4. Catalyst Performance
The water–gas shift activity was tested using a fixed bed reactor with a 4 mm inner diameter stainless steel tube reactor. The 0.15 g catalysts were loaded between two layers of quartz wool into a reactor, which was placed inside a tube furnace. The synthesized catalyst was reduced with 5%H
2/N
2 at 400 °C for 1 h prior to the water–gas shift activity measurement. The WGS activity test of all catalysts was operated in the temperature range of 150–600 °C. The feed gas consisted of 5% CO, 10% H
2O, and 85% N
2, with a total flow rate of 100 mL/min. The flow rate of CO and N
2 gas was controlled by mass flow controller, whereas H
2O was fed using a syringe pump. The outlet gasses were identified by an online Shimadzu GC-14B gas chromatographer equipped with a thermal conductivity detector. The column used in the gas chromatography was a Unibead C. The CO conversion was calculated using the following equation:
where
nCO,in and
nCO,out represent the molar flow rates of CO at the reactor inlet and outlet, respectively.
3. Results and Discussion
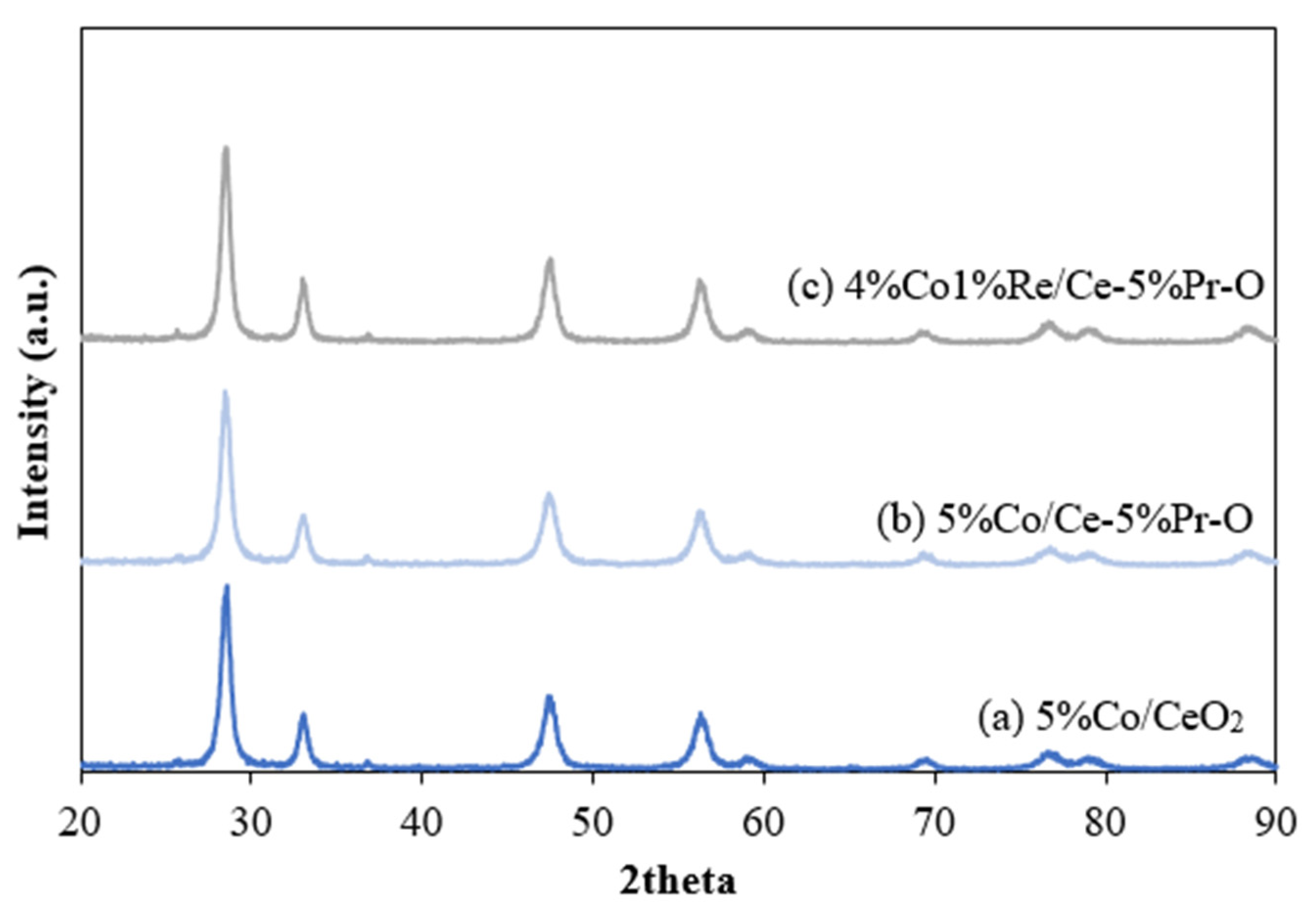
X-ray diffraction patterns analysis for cobalt-based catalysts (
Figure 1) provides the information on the structural characteristics and phase interactions within these catalysts. The XRD peaks of all samples correspond to JCPDS No. 43-1002 of standard CeO
2 fluorite structure. For the Co/Ce-Pr-O and CoRe/Ce-Pr-O catalysts, the absence of separate Pr
2O
3 phases suggests Pr incorporated into the ceria crystal structure as a solid solution. The observed leftward shift in the X-ray diffraction pattern towards lower 2θ angles indicates that cerium ions are substituted by smaller ions, such as Pr
2+. This ionic substitution induces distortion in the ceria unit cell and creates oxygen vacancies, which are crucial for enhancing catalytic activity by facilitating oxygen mobility and improving redox properties. The decrease in the 2θ angle was observed in the X-ray diffraction pattern of Co/Ce-Pr-O and CoRe/Ce-Pr-O catalysts when compared with Co/CeO
2 indicates the formation of oxygen vacancies in CeO
2, a key factor in enhancing the efficiency of the WGS reaction. The presence of oxygen vacancies promotes the reduction of CO to CO
2 by facilitating oxygen mobility and preventing carbon deposition, thereby improving the catalyst’s stability in this chemical process.
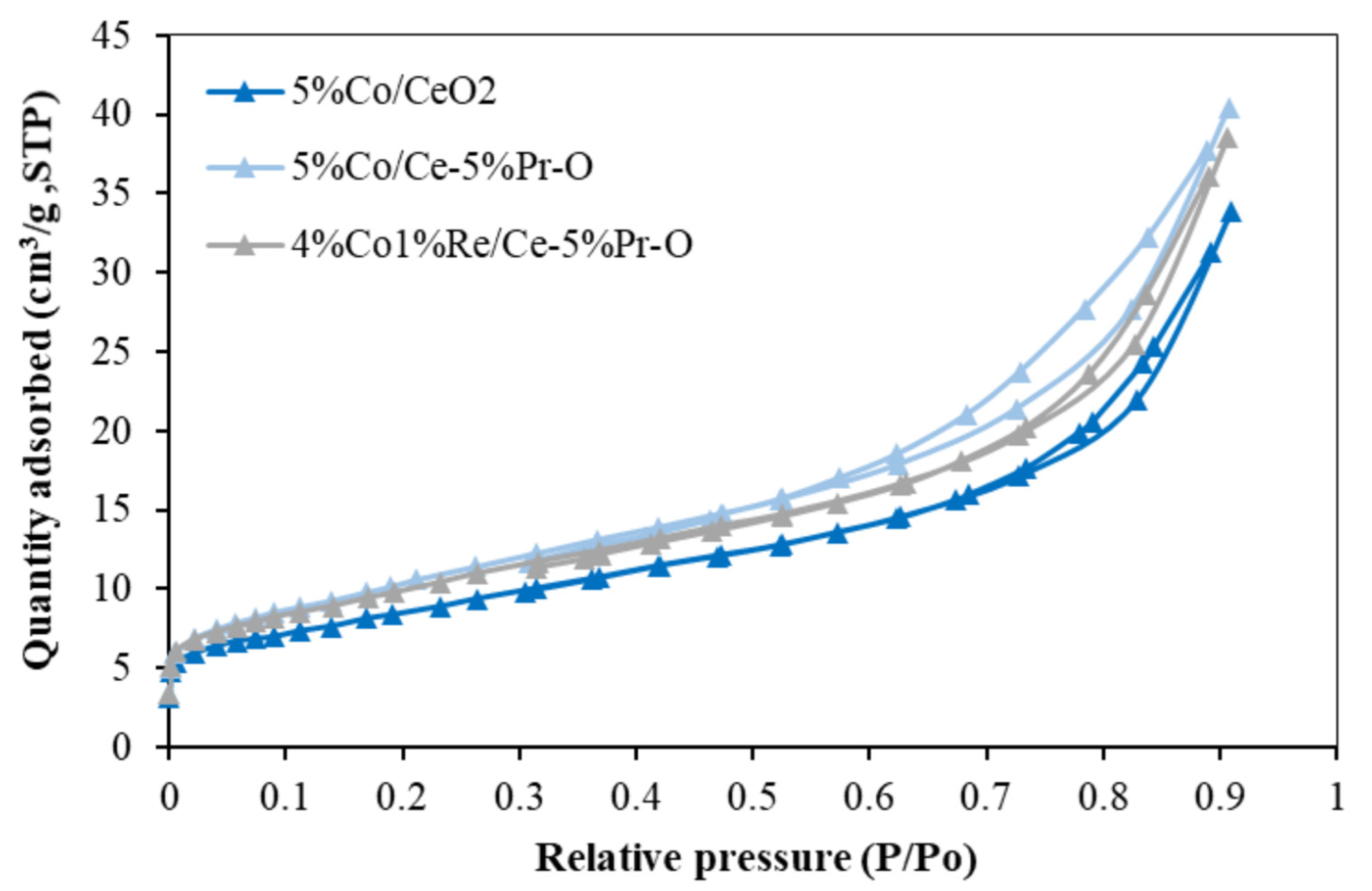
The synthesized catalysts demonstrated Type IV nitrogen adsorption–desorption isotherms accompanied by H3-type hysteresis loops at high relative pressures (P/Po > 0.8), as illustrated in
Figure 2. This behavior is indicative of the presence of mesoporous structures, which are characteristic of metal oxide-supported catalysts. Among the samples, 5%Co/Ce-5%Pr-O exhibited the highest nitrogen uptake across the entire relative pressure range, consistent with its superior BET surface area and porosity. The 4%Co1%Re/Ce-5%Pr-O catalyst also displayed enhanced adsorption behavior relative to 5%Co/CeO
2, suggesting that Re incorporation contributes positively to the development of surface accessible porosity. The observed H3-type hysteresis loops are typically associated with slit-like pores formed by the aggregation of plate-like particles, further supporting the presence of mesoporosity [
22]. This interpretation is corroborated by the average pore diameters presented in
Table 1, which fall within the mesoporous regime (2–50 nm). Furthermore, the incorporation of Pr was found to significantly increase the BET surface area and reduce the crystallite size, as observed for 5%Co/Ce-5%Pr-O, thereby indicating improved dispersion of cobalt species and suppression of particle growth during calcination. Although the addition of Re in the 4%Co1%Re/Ce-5%Pr-O catalyst slightly reduced the BET surface area compared to its Pr-only counterpart, it resulted in an increase in average pore diameter, implying a modification in pore architecture that may facilitate enhanced mass transport. These distinctions in textural characteristics are anticipated to have a pronounced impact on the catalytic behavior, particularly in reactions where diffusion and reactant accessibility to active sites are critical determinants of activity and selectivity.
The BET surface area was summarized in
Table 1. The incorporation of Pr into the 5%Co/CeO
2 catalyst increased the surface area while concurrently reducing the average pore diameter compared to Co supported on pure CeO
2. An increase in surface area of a catalyst results in enhancing the number of active sites for chemical reactions, thereby increasing the catalyst’s ability to accommodate reactant and product molecules. In addition to the BET surface area and pore characteristics, the crystallite size of the catalysts, as presented in
Table 1, provides further insight into the structural properties of the materials. Among the tested samples, the 5%Co/Ce–5%Pr–O catalyst exhibited the smallest crystallite size (10.66 nm), which correlates with its highest surface area (40 m
2/g) and smallest average pore diameter (6.44 nm). This observation suggests that the incorporation of Pr not only improves surface textural properties but also contributes to the inhibition of crystallite growth, leading to more dispersed active phases. In contrast, the 5%Co/Ce–5%Sm–O catalyst displayed the largest crystallite size (13.62 nm) and the lowest surface area (24 m
2/g), indicating a lower degree of active phase dispersion and potentially greater particle aggregation.
The morphology and elemental distribution of the as-synthesized catalysts were investigated using Scanning Electron Microscopy (SEM) coupled with Energy Dispersive X-ray Spectroscopy (EDS) for elemental mapping.
Figure 3,
Figure 4 and
Figure 5 present the SEM images and corresponding elemental maps for the 5%Co/CeO
2, 5%Co/CePrO, and 4%Co1%Re/CePrO catalysts, respectively. These catalysts exhibit an agglomerated particulate morphology, with finer particles dispersed among larger aggregates. Crucially, elemental maps for Ce, Pr, Co, and Re confirm their relatively uniform dispersion across the catalyst surface.
The consistent and relatively homogeneous distribution of metallic and support elements observed across all samples, particularly the active Co and promotional Re, indicates effective synthesis methods. This uniform dispersion is anticipated to positively influence catalytic performance by maximizing the accessibility of active sites for the water–gas shift (WGS) reaction.
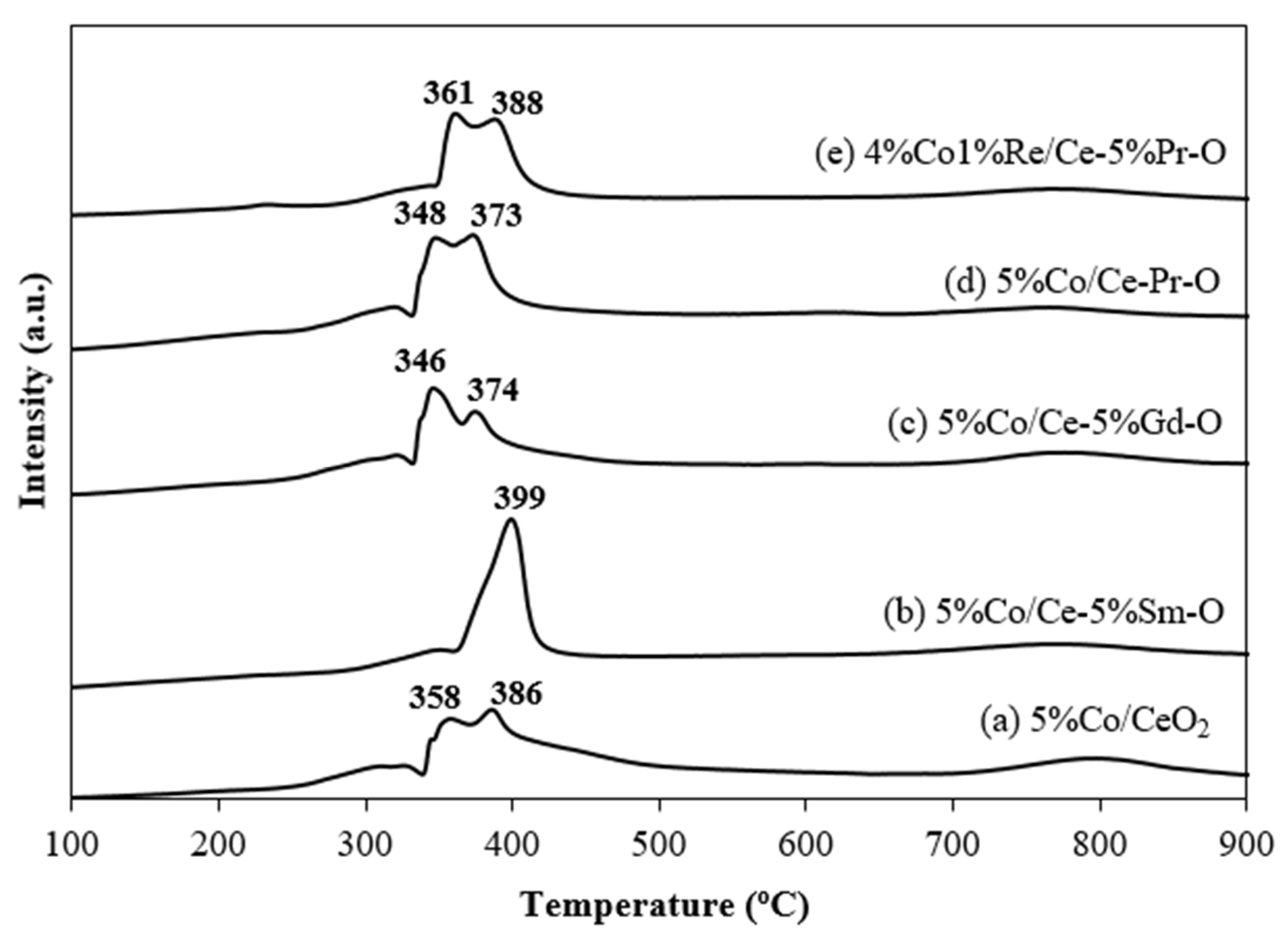
H
2-TPR profiles (
Figure 6) presents the reduction behavior of Co based catalysts. The primary reduction peaks observed at 346–399 °C correspond to the reduction of Co species interacting with surface ceria, while peaks in the range of 765–797 °C are assigned to the reduction in bulk ceria. Notably, the incorporation of rare earth promoters such as Pr and Gd induces a shift in the surface reduction peaks toward lower temperatures. For example, the Co/Ce-Pr-O catalyst exhibits reduction peaks at 348 and 373 °C, whereas the Co/CeO
2 catalyst shows corresponding peaks at 358 and 386 °C. This shift underscores the role of Pr in enhancing the reducibility of Co catalysts which leads to improve the catalyst’s efficiency in reduction reactions. Such enhanced reducibility directly benefits the water–gas shift reaction mechanism by promoting oxygen mobility and increasing oxygen vacancies.
The quantitative analysis of H
2 consumption of Co catalysts (
Table 2) was obtained by measuring the area of H
2-TPR curve. The H
2 consumption provides direct evidence for promoter-induced oxygen vacancy formation. For this work, the reduction peaks of cobalt species were only considered. The results show that 5%Co/Ce-5%Pr-O and 1%Re4%Co/Ce-5%Pr-O have higher H
2 consumption values than that of the 5%Co/CeO
2 catalyst.
The Raman spectra of the catalysts provide significant insights into the formation of oxygen vacancies. The characteristic F
2g mode at ~460 cm
−1, associated with the cubic fluorite structure of CeO
2, is clearly observed in all samples. However, the appearance of additional broad bands around ~590 cm
−1, typically attributed to oxygen vacancies or lattice defects, becomes increasingly prominent upon Pr doping and Co incorporation. As shown in
Figure 7, the Ce–5%Pr–O support exhibits enhanced oxygen vacancies (Vo) formation due to Pr
3+ substitution, while the 5%Co/Ce–5%Pr–O catalyst displays the most intense defect-related band, suggesting a synergistic effect between Co and Pr in generating oxygen vacancies. The enhanced formation of oxygen vacancies upon Pr doping, as evidenced by the increasing intensity of the defect-related Raman band at ~590 cm
−1, can be attributed to the substitution of Pr
3+ for Ce
4+ in the CeO
2 lattice. This aliovalent substitution introduces a charge imbalance, which is compensated by the generation of oxygen vacancies (Vo) to maintain overall electroneutrality. This structural modification not only explains the observed increase in the defect band in Raman spectra but also enhances the redox behavior critical for catalytic reactions.
Figure 8 illustrates the NH
3-TPD profiles of Co/Ce-Pr-O and CoRe/Ce-Pr-O catalysts. The NH
3-TPD peak at low temperature (below 200 °C) corresponds to weakly adsorbed NH
3 molecules, while the peak at high temperature represents strongly chemisorbed NH
3, indicating significant interactions between NH
3 and the catalyst surface. For 1%Re4%Co/Ce-5%Pr-O, a higher NH
3 desorption area is observed compared to 5%Co/Ce-5%Pr-O, indicating that the addition of Re enhances the surface acidity of the catalyst. This result is attributed to the enhancement of the number of active sites due to the surface acidity of the catalyst promotes interacting with the weak base of CO reactant. Therefore, an increase in surface acidity enhances CO adsorption, leading to improved catalytic activity in redox reactions.
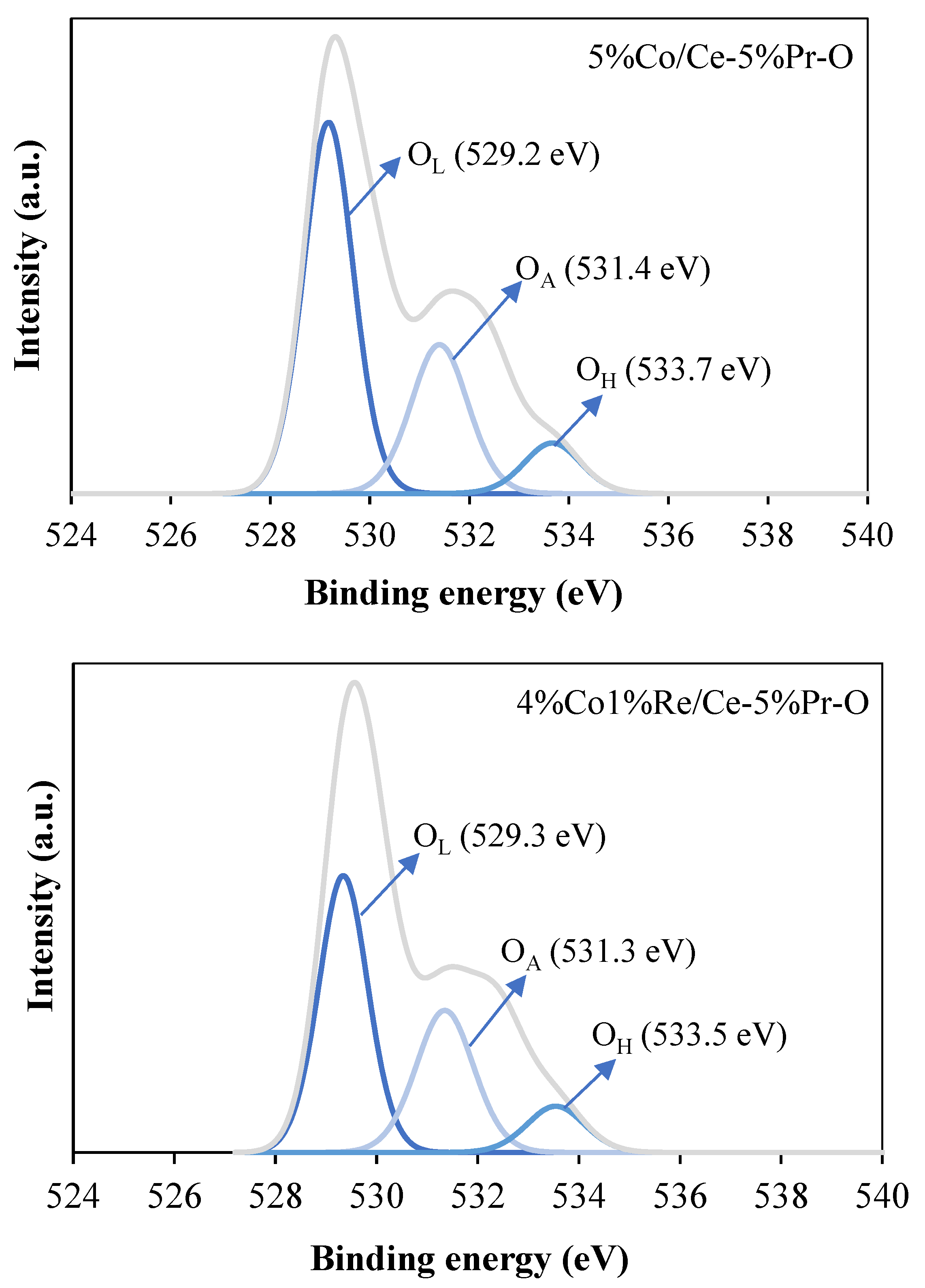
Figure 9 presents the O 1s XPS spectra for the (a) 5%Co/Ce-5%Pr-O and (b) 4%Co1%Re/Ce-5%Pr-O catalysts. Three distinct oxygen species were identified in all catalysts, with characteristic peaks at approximately 529 eV (O
L), 531 eV (O
A), and 533 eV (O
H), corresponding to lattice oxygen in metal oxide, chemically adsorbed oxygen on the catalyst surface, and hydroxyl oxygen species, respectively. The ratio of O
A/O
L serves as a significant indicator of catalytic activity, especially in catalysts used for oxidation reactions. A high O
A/O
L ratio serves as a reliable indicator of oxygen transfer and exchange dynamics on the catalyst surface, while the availability of highly mobile oxygen species mitigates coke formation. In the case of the 4%Co1%Re/Ce-5%Pr-O catalyst, the O
A/O
L ratio is 0.60, indicating a higher oxygen vacancy concentration compared to the 5%Co/Ce-5%Pr-O catalyst, which has an O
A/O
L ratio of 0.46. This result indicates that the 4%Co1%Re/Ce-5%Pr-O catalyst possesses enhanced potential for promoting reactions reliant on active oxygen species due to its higher oxygen vacancy concentration. The oxygen vacancies are critical for enhancing the redox properties and improving oxygen transfer capabilities. Therefore, the presence of rhenium stabilizes the Pr-containing lattice and promotes the formation of oxygen vacancies.
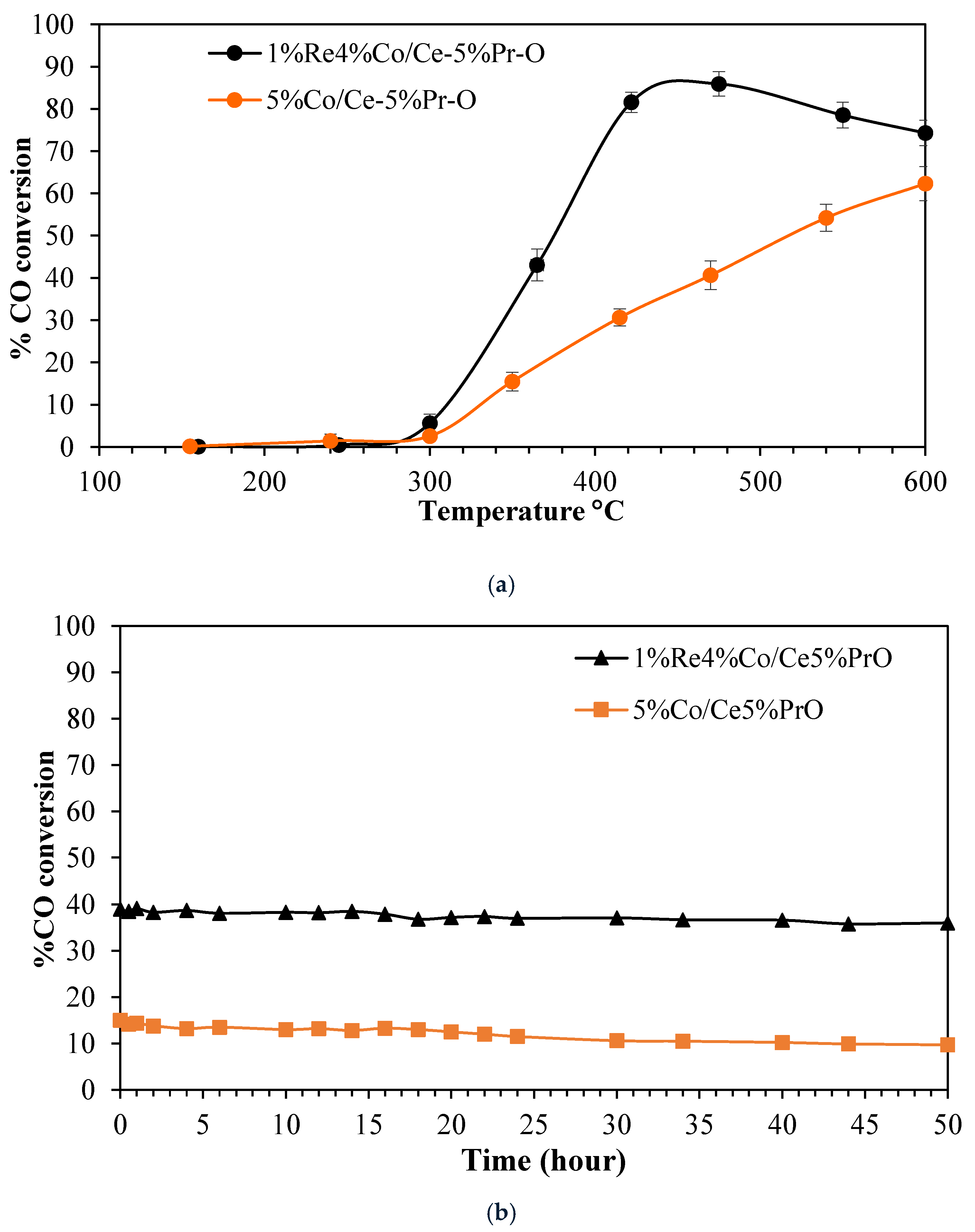
Figure 10a presents the catalytic performance of CoRe/Ce-Pr-O and Co/Ce-Pr-O catalysts for the water–gas shift reaction, demonstrating that the CoRe/Ce-Pr-O catalyst exhibits a markedly higher CO conversion efficiency when compared with Co/Ce-Pr-O. The superior catalytic activity of CoRe/Ce-Pr-O is due to the positive effect of Re, which not only enhances the acidity on the catalyst surface, leading to increased surface exposure of active sites, but also significantly increases the reducibility, oxygen mobility, and oxygen vacancies. The CoRe/Ce-Pr-O catalyst achieved a maximum CO conversion of 86% at about 450 °C, substantially outperforming the Co/Ce-Pr-O catalyst, which exhibited only 62% CO conversion at 600 °C. This observation indicates that the incorporation of rhenium effectively improves the catalytic performance of the WGS reaction. CoRe/Ce-Pr-O and Co/Ce-Pr-O were further examined at 350 °C for 50 h.
Figure 10b presents the CO conversion versus time during the stability test. The WGS activity of CoRe/Ce-Pr-O was kept constant for the whole period of 50 h, while a 6% decrease in WGS activity was found for Co/Ce-Pr-O catalyst. Therefore, CoRe/Ce-Pr-O catalyst is more resistant towards deactivation than Co/Ce-Pr-O. The addition of rhenium to Co/Ce-Pr-O catalysts enhanced the catalytic activity. This enhancement is attributed to strong metal–support interactions (SMSI). Specifically, rhenium can donate its electrons to the support material, creating oxygen vacancies which are crucial for WGS reaction mechanisms [
23].
The additions of praseodymium and rhenium to the Co/CeO2 catalyst for the water–gas shift (WGS) reaction have distinct effects. Pr acts as a dopant, modifying the bulk structure of ceria, while Re deposits on the surface, acting as a modifier of the surface properties. Praseodymium addition enhances the WGS activity by increasing surface area, reducing crystallite size and improving oxygen vacancy formation by replacing Ce4+ ions in the ceria lattice. On the other hand, Re can enhance the catalytic activity by interacting with Co to form bimetallic clusters which can be more active for the WGS reaction than the monometallic catalysts.
A comparison of the catalytic performance of the Co catalysts with similar articles is summarized in
Table 3. The catalytic activities of our synthesized Co-based catalysts, specifically 5%Co/Ce-Pr-O and 4%Co1%Re/Ce-Pr-O, were investigated. The support preparation method used in this work was the combustion method, a technique known for its simplicity, speed, and single-step synthesis approach. The 4%Co1%Re/Ce-Pr-O catalyst prepared by combustion exhibited a remarkably high CO conversion of 86% at 450 °C, which is highly competitive when compared to previously reported catalysts. For instance, a similar catalyst, 5%Co1%Re/CeO
2, prepared via urea gelation [
24], achieved a comparable conversion of 85% but the preparation process of this catalyst is more complicated. Therefore, considering the ease and rapidity of the combustion synthesis method employed in our study, the performance of our 4%Co1%Re/Ce-Pr-O catalyst is particularly noteworthy.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}