
In Silico Analysis of Vitamin D Interactions with Aging Proteins: Docking, Molecular Dynamics, and Solvation Free Energy Studies


 and
and
Abstract
1. Introduction
2. Computational Methodologies
2.1. Molecular Docking Calculations
2.1.1. Ligand Preparation
2.1.2. Protein Preparation
2.2. Molecular Dynamic (MD) Simulation
2.3. Binding Free Energy Calculated by Molecular Mechanics–Poisson–Boltzmann and Surface Area (MM-PBSA)
2.4. Solvation Free Energy Calculations
3. Results and Discussion
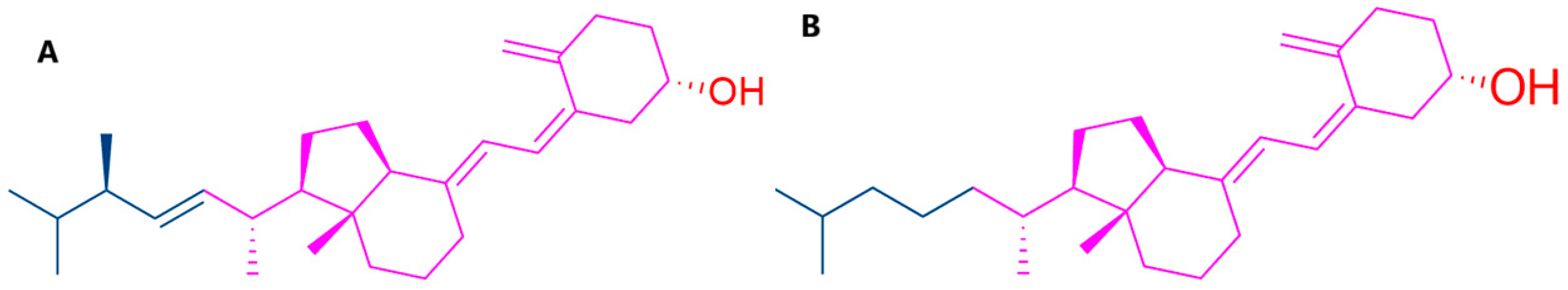
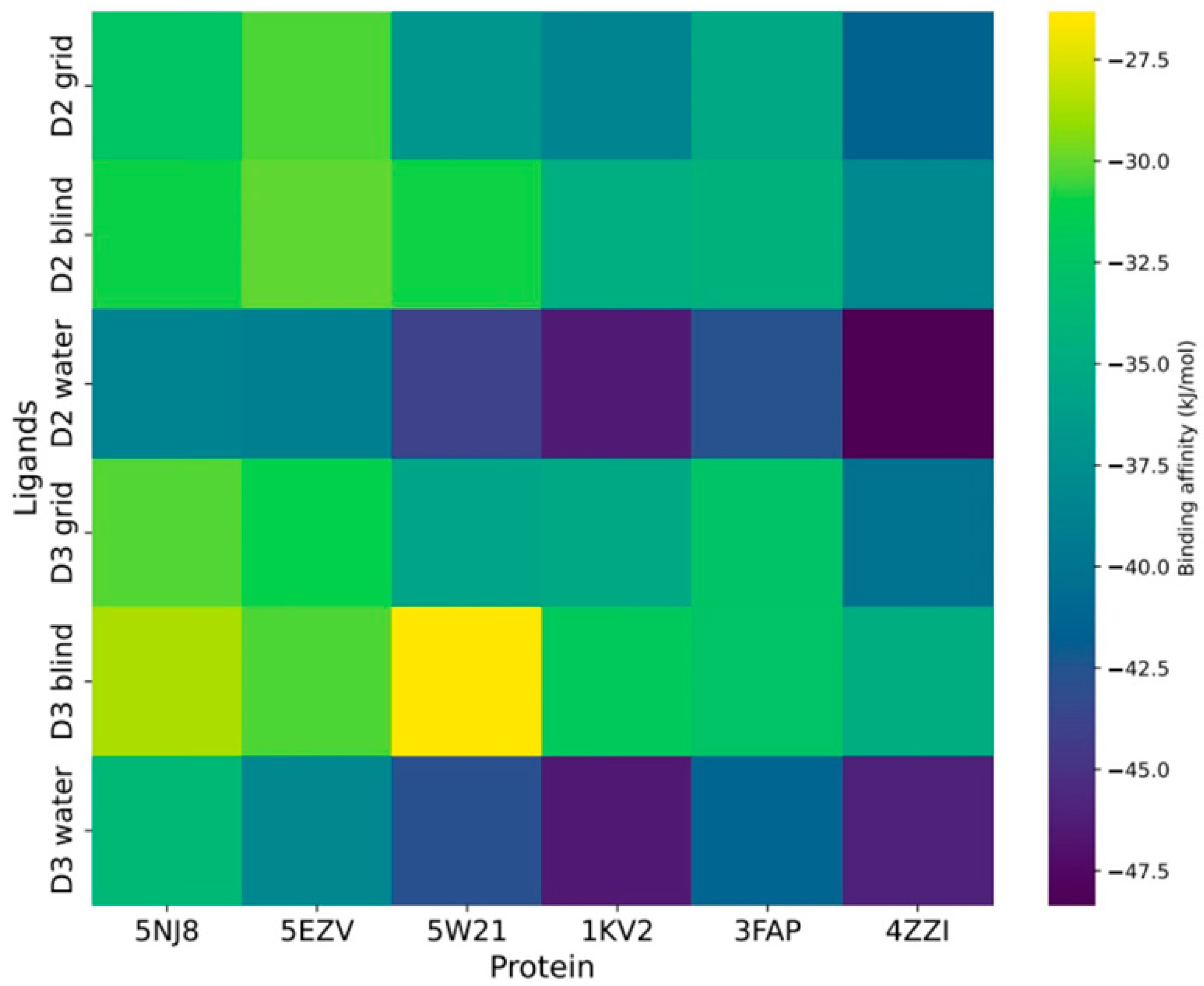
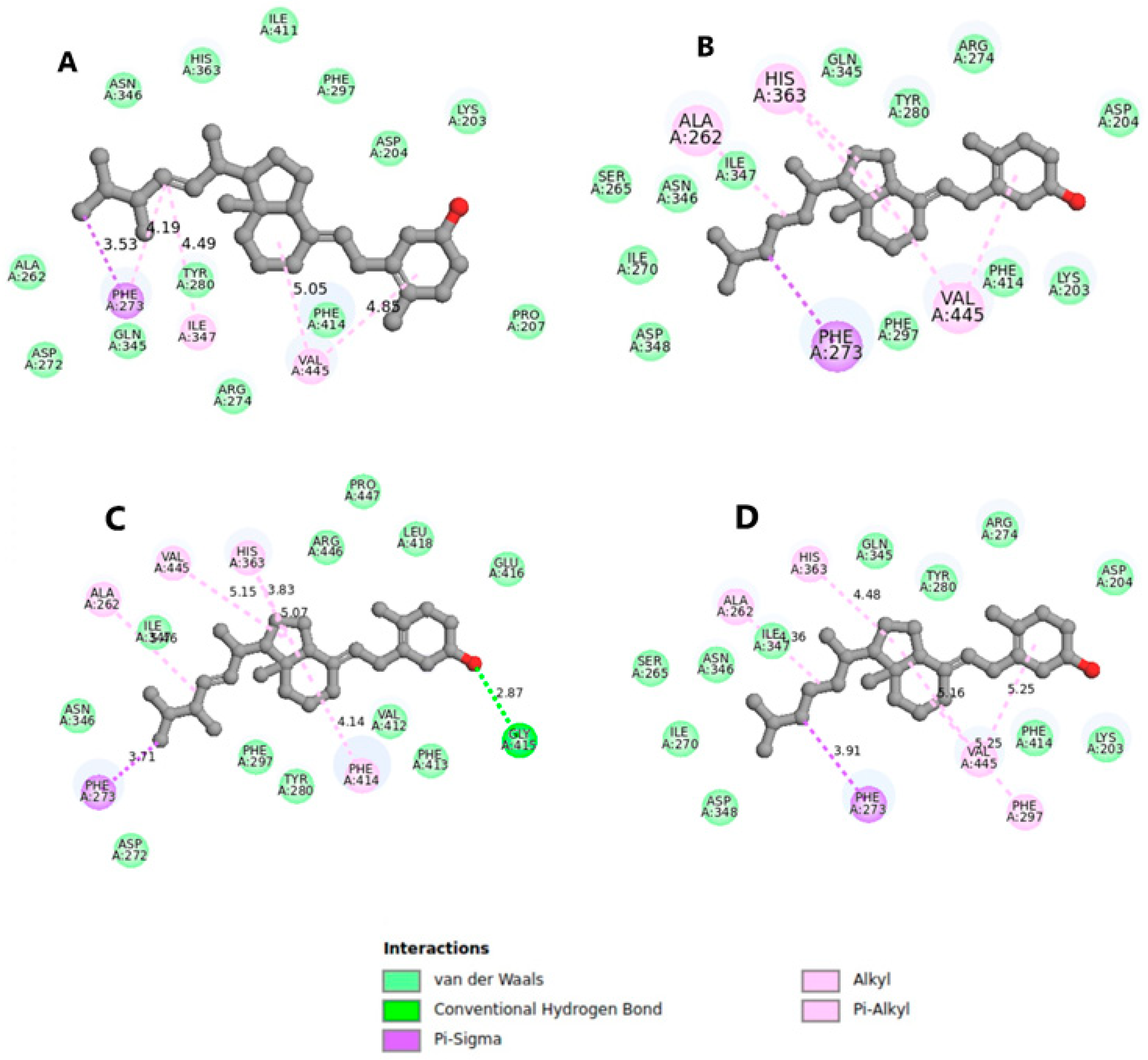
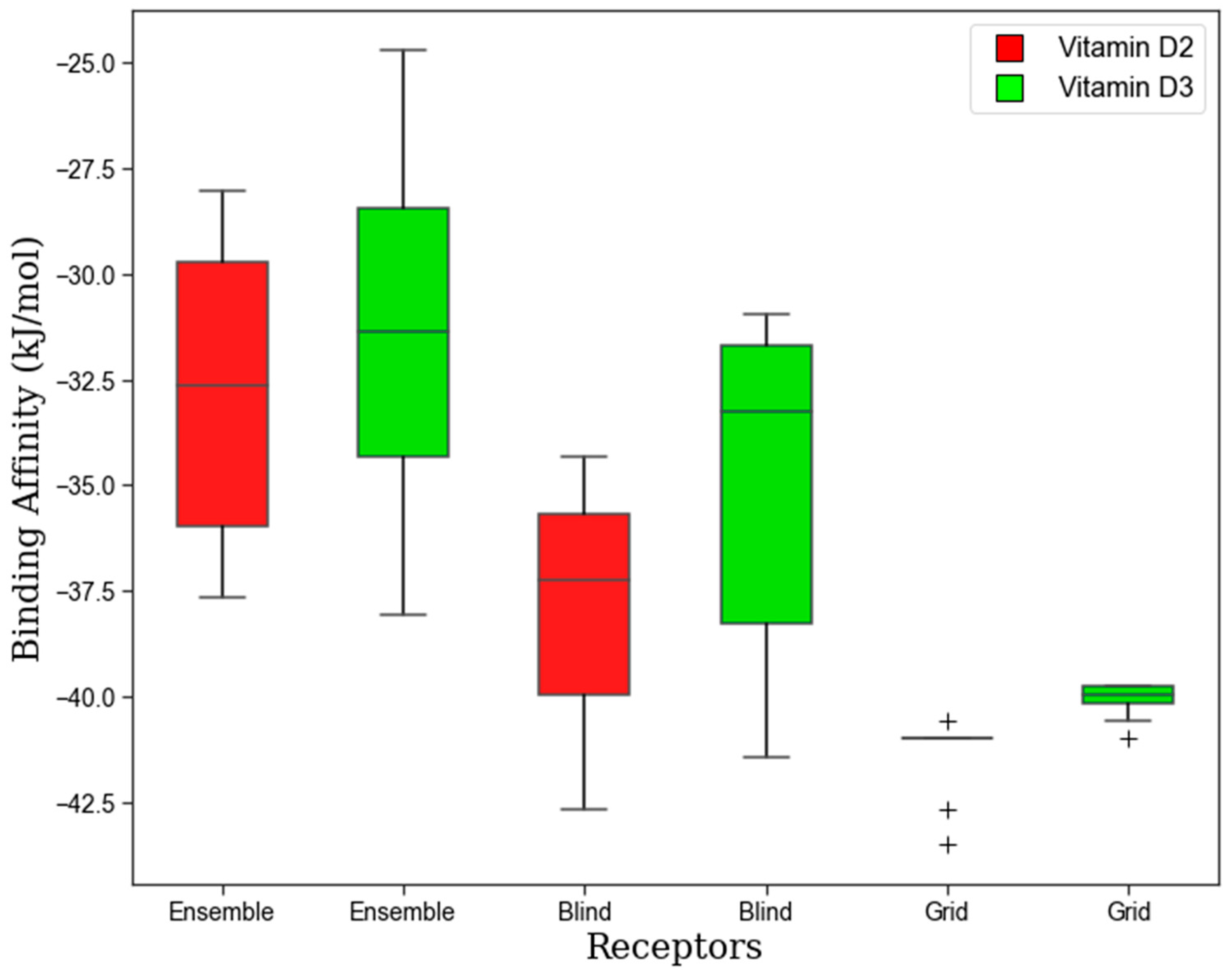
3.1. Molecular Interactions of Vitamin D with Key Aging-Related Proteins
Accommodating Receptor Flexibility: The Relaxed Complex Scheme (RCS)
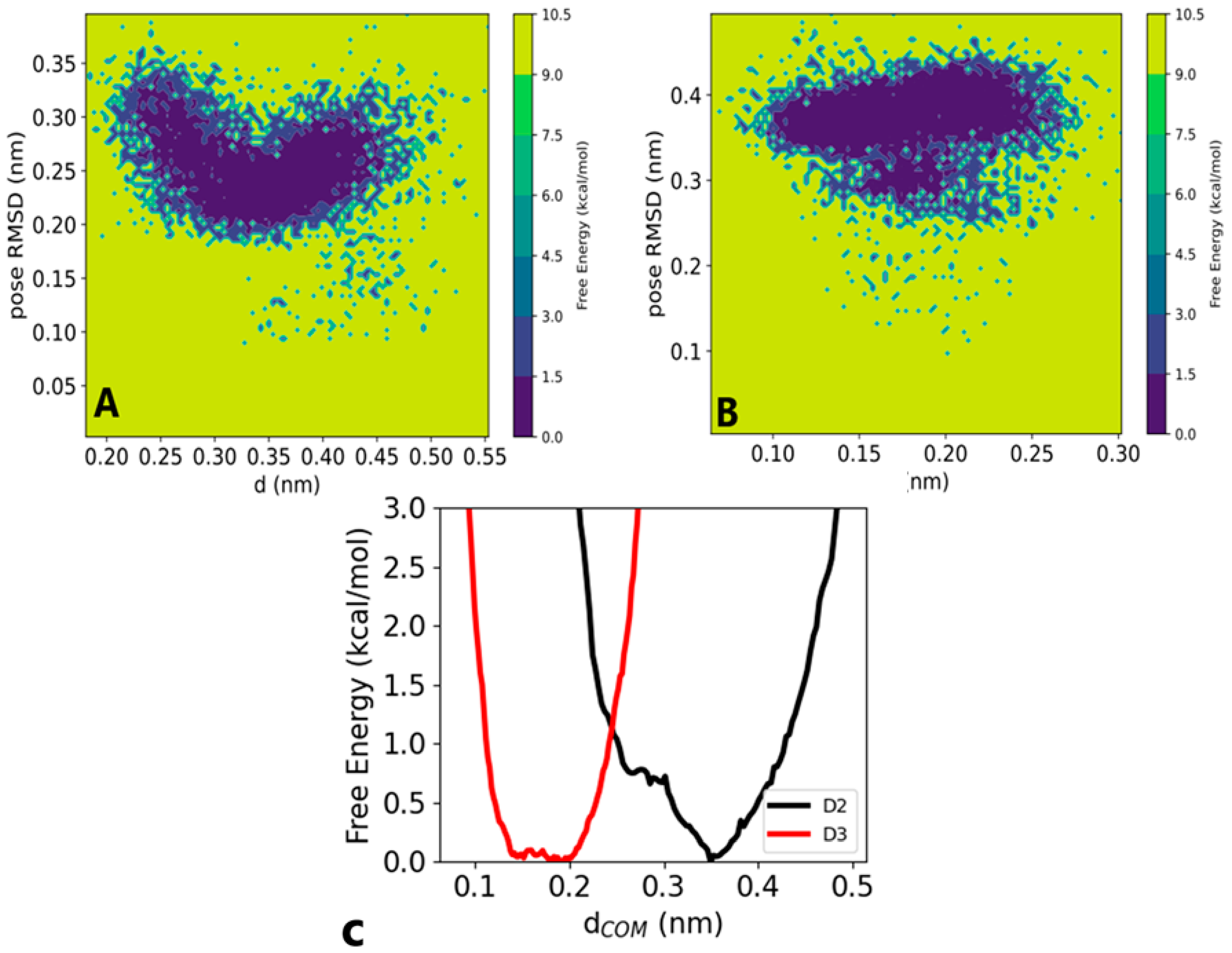
3.2. Molecular Dynamics Simulation
3.2.1. Structural Stability and Compactness
3.2.2. Vitamin D Fluctuation within Sirtuin 1 Receptor
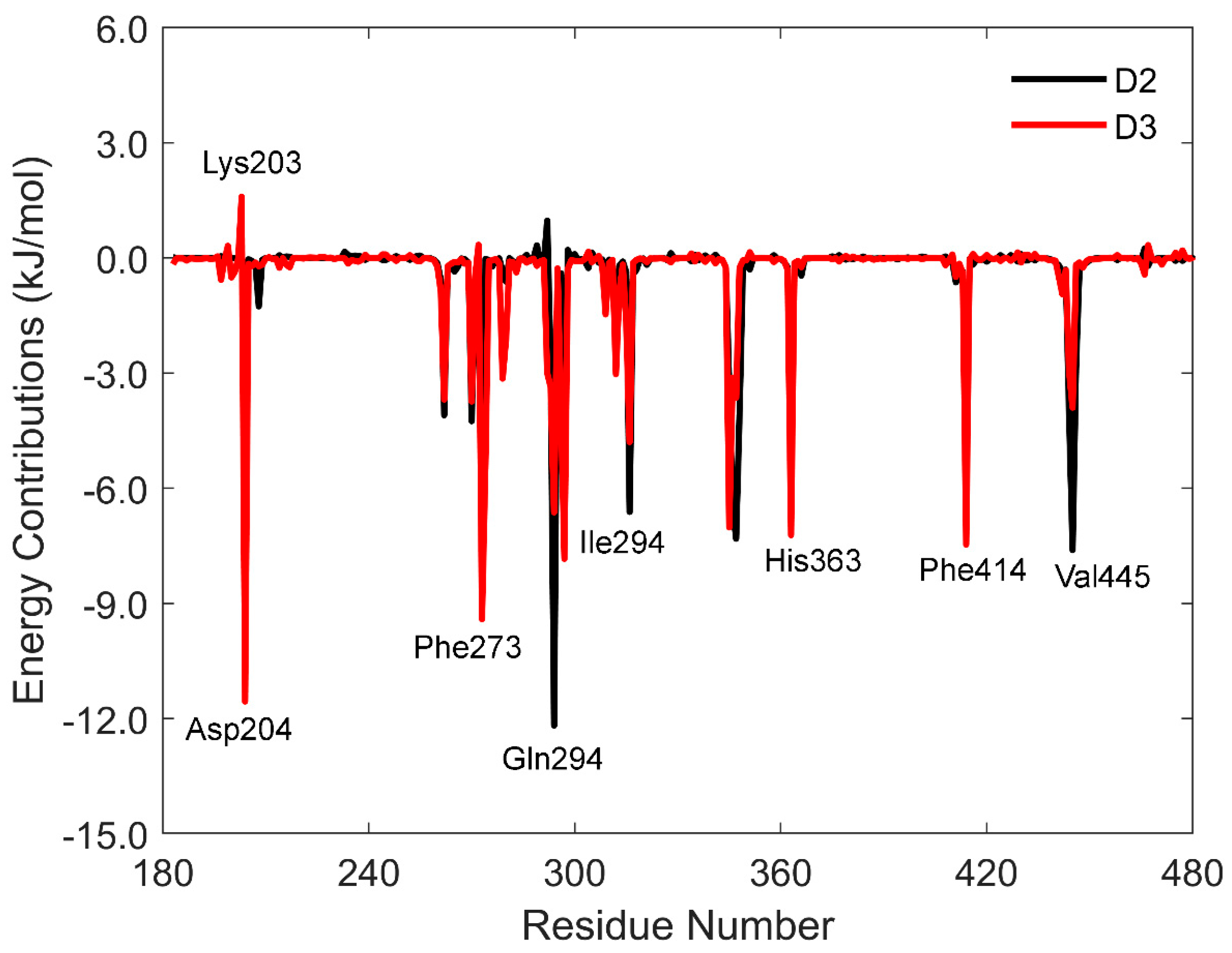
3.3. MM-PBSA Binding Free Energy Calculation
3.4. Solvation Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rando, T.A. Stem cells, ageing and the quest for immortality. Nature 2006, 441, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L. Stem cell aging. Mech. Ageing Dev. 2021, 193, 111394. [Google Scholar] [CrossRef]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar] [CrossRef]
- López-Otín, C.; Kroemer, G. Hallmarks of Health. Cell 2021, 184, 33–63. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Lages, A.; Lopes, V.; Horta, J.; Espregueira-Mendes, J.; Andrade, R.; Rebelo-Marques, A. Therapeutics That Can Potentially Replicate or Augment the Anti-Aging Effects of Physical Exercise. Int. J. Mol. Sci. 2022, 23, 9957. [Google Scholar] [CrossRef] [PubMed]
- Paredes-López, O.; Cervantes-Ceja, M.L.; Vigna-Pérez, M.; Hernández-Pérez, T. Berries: Improving Human Health and Healthy Aging, and Promoting Quality Life—A Review. Plant Foods Hum. Nutr. 2010, 65, 299–308. [Google Scholar] [CrossRef]
- Tosato, M.; Zamboni, V.; Ferrini, A.; Cesari, M. The aging process and potential interventions to extend life expectancy. Clin. Interv. Aging 2007, 2, 401–412. [Google Scholar]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- Russo, G.L.; Spagnuolo, C.; Russo, M.; Tedesco, I.; Moccia, S.; Cervellera, C. Mechanisms of aging and potential role of selected polyphenols in extending healthspan. Biochem. Pharmacol. 2020, 173, 113719. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.D.; Wagers, A.J. Stem cell aging: Mechanisms, regulators and therapeutic opportunities. Nat. Med. 2014, 20, 870–880. [Google Scholar] [CrossRef]
- Pignatti, C.; D’Adamo, S.; Stefanelli, C.; Flamigni, F.; Cetrullo, S. Nutrients and Pathways that Regulate Health Span and Life Span. Geriatrics 2020, 5, 95. [Google Scholar] [CrossRef]
- Fantini, C.; Corinaldesi, C.; Lenzi, A.; Migliaccio, S.; Crescioli, C. Vitamin D as a Shield against Aging. Int. J. Mol. Sci. 2023, 24, 4546. [Google Scholar] [CrossRef]
- Sosa-Díaz, E.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. The role of vitamin D on redox regulation and cellular senescence. Free Radic. Biol. Med. 2022, 193, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Żmijewski, M.A. Nongenomic Activities of Vitamin D. Nutrients 2022, 14, 5104. [Google Scholar] [CrossRef]
- Ekimoto, T.; Kudo, T.; Yamane, T.; Ikeguchi, M. Mechanism of Vitamin D Receptor Ligand-Binding Domain Regulation Studied by gREST Simulations. J. Chem. Inf. Model. 2021, 61, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.R.; Granic, A.; Aspray, T.J. Vitamin D and Ageing. In Biochemistry and Cell Biology of Ageing: Part I Biomedical Science; Harris, J.R., Korolchuk, V.I., Eds.; Springer: Singapore, 2018; pp. 191–220. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D deficiency accelerates ageing and age-related diseases: A novel hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef]
- Gallagher, J.C. Vitamin D and Aging. Endocrinol. Metab. Clin. 2013, 42, 319–332. [Google Scholar] [CrossRef]
- Hossein-nezhad, A.; Holick, M.F. Vitamin D for Health: A Global Perspective. Mayo Clin. Proc. 2013, 88, 720–755. [Google Scholar] [CrossRef]
- Meehan, M.; Penckofer, S. The Role of Vitamin D in the Aging Adult. J. Aging Gerontol. 2014, 2, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Salekeen, R.; Ahmed, A.; Islam, M.E.; Billah, M.M.; Rahman, H.; Islam, K.M.D. In-silico screening of bioactive phytopeptides for novel anti-ageing therapeutics. J. Biomol. Struct. Dyn. 2022, 40, 4475–4487. [Google Scholar] [CrossRef] [PubMed]
- Voltan, G.; Cannito, M.; Ferrarese, M.; Ceccato, F.; Camozzi, V. Vitamin D: An Overview of Gene Regulation, Ranging from Metabolism to Genomic Effects. Genes 2023, 14, 1691. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Bergadà, L.; Pallares, J.; Arcidiacono, M.V.; Cardus, A.; Santacana, M.; Valls, J.; Cao, G.; Fernàndez, E.; Dolcet, X.; Dusso, A.S.; et al. Role of local bioactivation of vitamin D by CYP27A1 and CYP2R1 in the control of cell growth in normal endometrium and endometrial carcinoma. Lab. Investig. 2014, 94, 608–622. [Google Scholar] [CrossRef]
- Fetahu, I.S.; Höbaus, J.; Kállay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Dastani, Z.; Li, R.; Richards, B. Genetic Regulation of Vitamin D Levels. Calcif. Tissue Int. 2013, 92, 106–117. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37 (Suppl. S2), W623–W633. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Roberts, B.C.; Mancera, R.L. Ligand–Protein Docking with Water Molecules. J. Chem. Inf. Model. 2008, 48, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Mohamed, M.; Zayed, M.; Gerlach, C.; Heine, A.; Hangauer, D.; Klebe, G. More than a Simple Lipophilic Contact: A Detailed Thermodynamic Analysis of Nonbasic Residues in the S1 Pocket of Thrombin. J. Mol. Biol. 2009, 390, 56–69. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Cheatham, T.E., III; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Darden, T.; Perera, L.; Li, L.; Pedersen, L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 7, R55–R60. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- White, J.A.; Román, F.L.; González, A.; Velasco, S. Periodic boundary conditions and the correct molecular-dynamics ensemble. Phys. A Stat. Mech. Its Appl. 2008, 387, 6705–6711. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Dill, K.; Bromberg, S.; Stigter, D. Polymer Solutions; Garland Science: New York, NY, USA, 2010; Volume 3, pp. 643–657. [Google Scholar]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef] [PubMed]
- Mey, A.; Allen, B.K.; Macdonald, H.E.B.; Chodera, J.D.; Hahn, D.F.; Kuhn, M.; Michel, J.; Mobley, D.L.; Naden, L.N.; Prasad, S.; et al. Best Practices for Alchemical Free Energy Calculations [Article v1.0]. Living J. Comput. Mol. Sci. 2020, 2, 18378. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Voityuk, A.A.; Vyboishchikov, S.F. A simple COSMO-based method for calculation of hydration energies of neutral molecules. Phys. Chem. Chem. Phys. 2019, 21, 18706–18713. [Google Scholar] [CrossRef]
- Voityuk, A.A.; Vyboishchikov, S.F. Fast and accurate calculation of hydration energies of molecules and ions. Phys. Chem. Chem. Phys. 2020, 22, 14591–14598. [Google Scholar] [CrossRef] [PubMed]
- Vyboishchikov, S.F.; Voityuk, A.A. Fast non-iterative calculation of solvation energies for water and non-aqueous solvents. J. Comput. Chem. 2021, 42, 1184–1194. [Google Scholar] [CrossRef]
- Paul, L.; Namba-Nzanguim, C.T.; Telesphory, A.; Oppong Mensah, J.; Mteremko, D.; Costa, R.; Katundu, S.M.; Kwiyukwa, L.P.; Kambaine, N.D.; Juvenary, J.; et al. Investigation of the structure, stability, and relative solubility of psilocybin in water and pure organic solvents: A molecular simulation study. J. Mol. Liq. 2023, 392, 123479. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef]
- Geidl, S.; Bouchal, T.; Raček, T.; Svobodová Vařeková, R.; Hejret, V.; Křenek, A.; Abagyan, R.; Koča, J. High-quality and universal empirical atomic charges for chemoinformatics applications. J. Cheminformatics 2015, 7, 59. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Hutchison, G.R. Fast, efficient fragment-based coordinate generation for Open Babel. J. Cheminformatics 2019, 11, 49. [Google Scholar] [CrossRef]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Vyboishchikov, S.F. A quick solvation energy estimator based on electronegativity equalization. J. Comput. Chem. 2023, 44, 307–318. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.-C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Onuchic, J.N. Water and proteins: A love–hate relationship. Proc. Natl. Acad. Sci. USA 2004, 101, 3325–3326. [Google Scholar] [CrossRef]
- Schiebel, J.; Gaspari, R.; Wulsdorf, T.; Ngo, K.; Sohn, C.; Schrader, T.E.; Cavalli, A.; Ostermann, A.; Heine, A.; Klebe, G. Intriguing role of water in protein-ligand binding studied by neutron crystallography on trypsin complexes. Nat. Commun. 2018, 9, 3559. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.; Patonai, A.; Simon-Szabó, L.; Takács, I. Interplay of Vitamin D and SIRT1 in Tissue-Specific Metabolism—Potential Roles in Prevention and Treatment of Non-Communicable Diseases Including Cancer. Int. J. Mol. Sci. 2023, 24, 6154. [Google Scholar] [CrossRef] [PubMed]
- Piwpong, R. Systematic Review on Anti-Aging Health Care. Naresuan Univ. J. Sci. Technol. (NUJST) 2018, 26, 98–112. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin Catalysis and Regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Evdw | Eelect | Epolar | Enonpolar | ΔGbinding |
|---|---|---|---|---|---|
| D2–Sirt1 | −211.28 ± 16.09 | −5.27 ± 9.88 | 97.92 ± 15.266 | −25.43 ± 1.13 | −144.07 ± 19.45 |
| D3–Sirt1 | −211.85 ± 12.41 | −36.8 ± 13.13 | 108.20 ± 16.34 | −26.66 ± 1.00 | −167.12 ± 16.35 |
| Solvent | ΔGsolv (Electrostatic) | ΔGsolv (LJ) | ΔGsolv (Total) |
|---|---|---|---|
| Water | −30.05 ± 0.05 | 18.72 ± 0.33 | −11.34 ± 0.33 |
| Ethanol | −18.52 ± 0.05 | −43.48 ± 0.21 | −62.01 ± 0.21 |
| Butanone | −13.15 ± 0.03 | −64.85 ± 0.20 | −78.00 ± 0.20 |
| Cyclohexane | −5.61 ± 0.03 | −78.06 ± 0.24 | −83.67 ± 0.24 |
| Solvent | ΔGsolv (Electrostatic) | ΔGsolv (LJ) | ΔGsolv (Total) |
|---|---|---|---|
| Water | −27.21 ± 0.05 | 18.06 ± 0.30 | −9.15 ± 0.30 |
| Ethanol | −18.26 ± 0.05 | −43.20 ± 0.20 | −61.46 ± 0.21 |
| Butanone | −13.95 ± 0.03 | −64.50 ± 0.19 | −78.46 ± 0.19 |
| Cyclohexane | −5.85 ± 0.02 | −77.29 ± 0.22 | −83.14 ± 0.23 |
| Solvent | ΔGsolv (Electrostatic) | ΔGsolv (Correction) | ΔGsolv (Total) |
|---|---|---|---|
| Water | −39.807 | 26.133 | −13.677 |
| Ethanol | −38.702 | −41.530 | −80.228 |
| Butanone | −38.112 | −83.467 | −121.579 |
| Cyclohexane | −16.288 | −79.940 | −96.228 |
| Solvent | ΔGsolv (Electrostatic) | ΔGsolv (Correction) | ΔGsolv (Total) |
|---|---|---|---|
| Water | −38.522 | 6.079 | −32.443 |
| Ethanol | −37.447 | −44.313 | −81.760 |
| Butanone | −36.882 | −80.768 | −117.650 |
| Cyclohexane | −15.761 | −76.224 | −91.985 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuntufye, E.; Paul, L.; Raymond, J.; Chacha, M.; Paluch, A.S.; Shadrack, D.M. In Silico Analysis of Vitamin D Interactions with Aging Proteins: Docking, Molecular Dynamics, and Solvation Free Energy Studies. ChemEngineering 2024, 8, 104. https://doi.org/10.3390/chemengineering8050104
Tuntufye E, Paul L, Raymond J, Chacha M, Paluch AS, Shadrack DM. In Silico Analysis of Vitamin D Interactions with Aging Proteins: Docking, Molecular Dynamics, and Solvation Free Energy Studies. ChemEngineering. 2024; 8(5):104. https://doi.org/10.3390/chemengineering8050104
Chicago/Turabian StyleTuntufye, Edna, Lucas Paul, Jofrey Raymond, Musa Chacha, Andrew S. Paluch, and Daniel M. Shadrack. 2024. "In Silico Analysis of Vitamin D Interactions with Aging Proteins: Docking, Molecular Dynamics, and Solvation Free Energy Studies" ChemEngineering 8, no. 5: 104. https://doi.org/10.3390/chemengineering8050104
APA StyleTuntufye, E., Paul, L., Raymond, J., Chacha, M., Paluch, A. S., & Shadrack, D. M. (2024). In Silico Analysis of Vitamin D Interactions with Aging Proteins: Docking, Molecular Dynamics, and Solvation Free Energy Studies. ChemEngineering, 8(5), 104. https://doi.org/10.3390/chemengineering8050104