Dynamically Operated Fischer-Tropsch Synthesis in PtL-Part 1: System Response on Intermittent Feed



Abstract
1. Introduction
1.1. Energy Transition and Technologies that May Be Required
1.2. Microstructured Reactor Technology
- What knowledge is needed for controlling a dynamic process in small-to-medium-size businesses?
- Can all different process steps be aligned in dynamic operation? What are their general limitations?
- What is the potential to reduce storages through dynamic synthesis?
- What are considerable “dynamic time periods” and ramping scenarios that plants need to tolerate?
- Can the prerequisites from “dynamic time periods” be met by reactors and/or plants?
- How is overall higher efficiency correlated to values of buffer reduction?
2. Materials and Methods
2.1. Analysis in Transient and Steady-State Operation
- A Diluter 004 that dilutes the sample with Ar to avoid detector overloading (below 500,000 counts s−1). Measurements were performed at 1:20 up to 1:30 dilution. Pressure fluctuation from 0.02–0.5 MPa can be compensated.
- An Airsense 2000 unit that can apply three different gases (or mixtures thereof) with different ionization energies: Hg (10 eV), Xe (12 eV) and Kr (14 eV); these gases can be switched within several milliseconds. Electron impact ionization is used to generate primary ions; those ions hit sample molecules and transfer their charge. A quadrupole high-frequency mass filter then separates gaseous molecules based on their mass number (0–500 amu) while the sample can be continuously measured with an impact detector using pulse counting electronics. Depending on the switching times of the quadrupole, the ionization gas and the required number of signals, the interval for a new measurement can range between < 1 s and several seconds. For detecting CO, CO2, CH4, and C3H8, approximately 3 s were needed to apply a mixture of Xe and Kr.
- An H-Sense unit where the sample is ionized via electron impact ionization so the concentration of H2 can be measured in response times below 300 ms.
2.2. Experimental Test Rig
- (1)
- a gas feeding system with mass flow controllers (MFCs; Brooks, NL),
- (2)
- a microstructured pilot scale reactor with evaporation cooling in patented design (Figure 3) and a reaction volume of approx. 150 cm3 designed to produce around 7 L of liquid product per day, depending on the process conditions; it was filled with 120 g commercial catalyst with 20 wt.% CO on optimized alumina,
- (3)
- a hot trap (HT) and cold trap (CT) to capture the different product phases; a micro-heat exchanger (µHE) cools down the stream between both traps (Figure 4).
2.3. Residence Time Distribution and Dead Time Measurements (Non-Reactive and Reactive Mode)
2.4. Quick Liquid and Gas Sampling (QS)
2.5. Dynamic Profiles by Oscillation of Feed Concentration
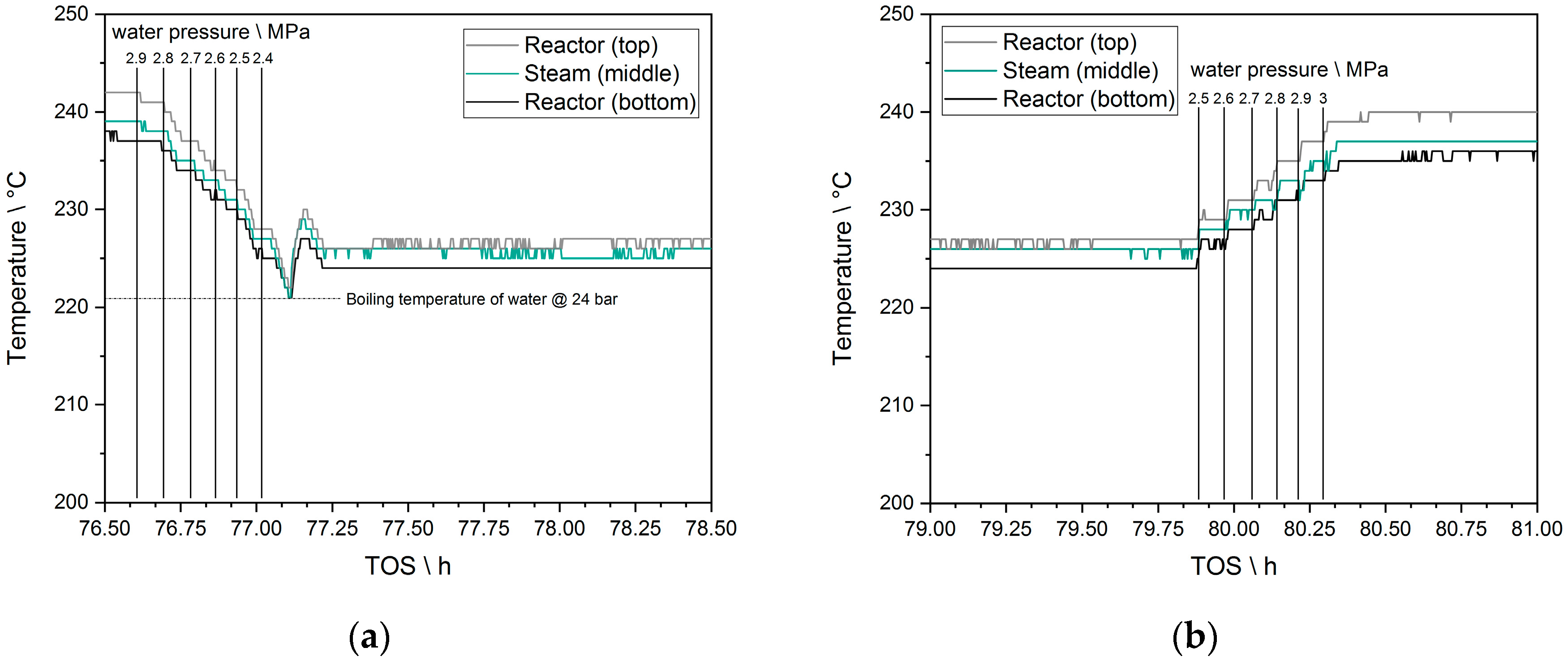
2.6. Evaporation Cooling as a Tool for Influencing the Reactor Temperature
3. Results and Discussion
3.1. General Product Composition and Comparison to Quick Sampling
3.2. Steady-State Compositions at Low and High H2/CO Conditions
3.3. RTD Measurements—None-Reactive Mode
3.4. RTD Measurements—Reactive Mode
3.5. Cycle Experiments—Concentration Switching
3.6. Cycle Experiments—Temperature Switching
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischer, F.; Tropsch, H. The preparation of synthetic oil mixtures (synthol) from carbon monoxide and hydrogen. Brennst. Chem. 1923, 4, 276–285. [Google Scholar]
- Dry, M.E. The Fischer-Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- De Klerk, A. Fischer-Tropsch Refining; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 9783527635603. [Google Scholar]
- Khorashadizadeh, M.; Atashi, H.; Mirzaei, A.A. Process conditions effects on Fischer-Tropsch product selectivity: Modeling and optimization through a time and cost-efficient scenario using a limited data size. J. Taiwan Inst. Chem. Eng. 2017, 80, 709–719. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and theoretical aspects of the catalytic Fischer-Tropsch process. Appl. Catal. A Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Maitlis, P.M.; De Klerk, A. Greener Fischer-Tropsch Processes for Fuels and Feedstocks; Wiley-VCH: Weinheim, Germany, 2013; ISBN 3527329455. [Google Scholar]
- Lu, Y.; Lee, T. Influence of the feed gas composition on the Fischer-Tropsch synthesis in commercial operations. J. Nat. Gas Chem. 2007, 16, 329–341. [Google Scholar] [CrossRef]
- Fundamentals of Industrial Catalytic Processes, 2nd ed.; Bartholomew, C.H., Farrauto, R.J., Eds.; Wiley-Interscience: Hoboken, NJ, USA, 2010; ISBN 9780471730071. [Google Scholar]
- Todic, B.; Nowicki, L.; Nikacevic, N.; Bukur, D.B. Fischer-Tropsch synthesis product selectivity over an industrial iron-based catalyst: Effect of process conditions. Catal. Today 2016, 261, 28–39. [Google Scholar] [CrossRef]
- Myrstad, R.; Eri, S.; Pfeifer, P.; Rytter, E.; Holmen, A. Fischer-Tropsch synthesis in a microstructured reactor. Catal. Today 2009, 147, S301–S304. [Google Scholar] [CrossRef]
- Piermartini, P.; Boeltken, T.; Selinsek, M.; Pfeifer, P. Influence of channel geometry on Fischer-Tropsch synthesis in microstructured reactors. Chem. Eng. J. 2017, 313, 328–335. [Google Scholar] [CrossRef]
- Sun, C.; Luo, Z.; Choudhary, A.; Pfeifer, P.; Dittmeyer, R. Influence of the condensable hydrocarbons on an integrated Fischer-Tropsch synthesis and hydrocracking process: Simulation and experimental validation. Ind. Eng. Chem. Res. 2017, 56, 13075–13085. [Google Scholar] [CrossRef]
- Sun, C.; Zhan, T.; Pfeifer, P.; Dittmeyer, R. Influence of Fischer-Tropsch synthesis (FTS) and hydrocracking (HC) conditions on the product distribution of an integrated FTS-HC process. Chem. Eng. J. 2017, 310, 272–281. [Google Scholar] [CrossRef]
- Sun, C. Direct Syngas-to-Fuel: Integration of Fischer—Tropsch Synthesis and Hydrocracking in Micro—Structured Reactors. Ph.D. Thesis, Karlsruhe Institute of Technology, Karlsruhe, Germany, 2017. [Google Scholar]
- Loewert, M.; Hoffmann, J.; Piermartini, P.; Selinsek, M.; Dittmeyer, R.; Pfeifer, P. Microstructured Fischer-Tropsch reactor scale-up and opportunities for decentralized application. Chem. Eng. Technol. 2019, 42, 2202–2214. [Google Scholar] [CrossRef]
- Adesina, A.A. Hydrocarbon synthesis via Fischer-Tropsch reaction: Travails and triumphs. Appl. Catal. A Gen. 1996, 138, 345–367. [Google Scholar] [CrossRef]
- Sousa, B.V.D.; Rodrigues, M.G.F.; Cano, L.A.; Cagnoli, M.V.; Bengoa, J.F.; Marchetti, S.G.; Pecchi, G. Study of the effect of cobalt content in obtaining olefins and paraffins using the Fischer-Tropsch reaction. Catal. Today 2011, 172, 152–157. [Google Scholar] [CrossRef]
- Dry, M.E. Commercial conversion of carbon monoxide to fuels and chemicals. J. Organomet. Chem. 1989, 372, 117–128. [Google Scholar] [CrossRef]
- Rausch, A.K.; Schubert, L.; Henkel, R.; Van Steen, E.; Claeys, M.; Roessner, F. Enhanced olefin production in Fischer-Tropsch synthesis using ammonia containing synthesis gas feeds. Catal. Today 2016, 275, 94–99. [Google Scholar] [CrossRef]
- Le Quéré, C.; Andrew, R.M.; Friedlingstein, P.; Sitch, S.; Hauck, J.; Pongratz, J.; Pickers, P.A.; Korsbakken, J.I.; Peters, G.P.; Canadell, J.G.; et al. Global carbon budget 2018. Earth Syst. Sci. Data 2018, 10, 2141–2194. [Google Scholar] [CrossRef]
- Horvath, S.; Fasihi, M.; Breyer, C. Techno-economic analysis of a decarbonized shipping sector: Technology suggestions for a fleet in 2030 and 2040. Energy Convers. Manag. 2018, 164, 230–241. [Google Scholar] [CrossRef]
- Kousoulidou, M.; Lonza, L. Biofuels in aviation: Fuel demand and CO2 emissions evolution in Europe toward 2030. Transport. Res. D 2016, 46, 166–181. [Google Scholar] [CrossRef]
- Guandalini, G.; Robinius, M.; Grube, T.; Campanari, S.; Stolten, D. Long-term power-to-gas potential from wind and solar power: A country analysis for Italy. Int. J. Hydrog. Energy 2017, 42, 13389–13406. [Google Scholar] [CrossRef]
- Vázquez, F.V.; Koponen, J.; Ruuskanen, V.; Bajamundi, C.; Kosonen, A.; Simell, P.; Ahola, J.; Frilund, C.; Elfving, J.; Reinikainen, M.; et al. Power-to-X technology using renewable electricity and carbon dioxide from ambient air: SOLETAIR proof-of-concept and improved process concept. J. CO2 Util. 2018, 28, 235–246. [Google Scholar] [CrossRef]
- International Energy Agency (IEA). CO2 Emissions from Fuel Combustion—Highlights. Available online: https://www.iea.org/reports/co2-emissions-from-fuel-combustion-2019 (accessed on 27 March 2020).
- Wagemann, K.; Ausfelder, F. White Paper E-Fuels—Mehr Als Eine Option—Finale Version; DECHEMA e.V.: Frankfurt a. M., Germany, 2017; ISBN 978-3-89746-198-7. [Google Scholar]
- SAPEA—Science Advice for Policy by European Academies. Novel Carbon Capture and Utilisation Technologies; SAPEA: Berlin, Germany, 2018; ISBN 9789279820076. [Google Scholar]
- Deutsche Energie-Argentur (DENA). Study: Integrated Energy Transition; Deutsche Energie-Agentur GmbH (dena): Berlin, Germany, 2018. [Google Scholar]
- European University Association. Energy transition and the future of energy research, innovation and education: An action agenda for European universities. Int. J. Prod. Res. 2017, 53, 59. [Google Scholar]
- IRENA. Global Energy Transformation: A Roadmap to 2050. Available online: https://irena.org/publications/2018/Apr/Global-Energy-Transition-A-Roadmap-to-2050 (accessed on 27 March 2020).
- IPCC. Global Warming of 1.5 °C An IPCC Special Report on the Impacts of Global Warming of 1.5 °C Above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change. Available online: https://www.ipcc.ch/sr15/ (accessed on 27 March 2020).
- Tremel, A. Electricity-Based Fuels; Springer International Publishing: Basel, Switzerland, 2018; ISBN 9783319724584. [Google Scholar]
- International Energy Agency (IEA). World Energy Outlook 2016. Available online: https://www.iea.org/reports/world-energy-outlook-2016 (accessed on 27 March 2020).
- Vaillancourt, K.; Bahn, O.; Roy, P.O.; Patreau, V. Is there a future for new hydrocarbon projects in a decarbonizing energy system? A case study for Quebec (Canada). Appl. Energy 2018, 218, 114–130. [Google Scholar] [CrossRef]
- Ridjan, I.; Mathiesen, B.V.; Connolly, D. Terminology used for renewable liquid and gaseous fuels based on the conversion of electricity: A review. J. Clean. Prod. 2016, 112, 3709–3720. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef]
- Reuß, M.; Grube, T.; Robinius, M.; Preuster, P.; Wasserscheid, P.; Stolten, D. Seasonal storage and alternative carriers: A flexible hydrogen supply chain model. Appl. Energy 2017, 200, 290–302. [Google Scholar] [CrossRef]
- Kotzur, L.; Markewitz, P.; Robinius, M.; Stolten, D. Time series aggregation for energy system design: Modeling seasonal storage. Appl. Energy 2018, 213, 123–135. [Google Scholar] [CrossRef]
- Brynolf, S.; Taljegard, M.; Grahn, M.; Hansson, J. Electrofuels for the transport sector: A review of production costs. Renew. Sustain. Energy Rev. 2018, 81, 1887–1905. [Google Scholar] [CrossRef]
- Leonard, G.; Francois-Lavet, V.; Ernst, D.; Meinrenken, C.J.; Lackner, K.S. Electricity storage with liquid fuels in a zone powered by 100% variable renewables. In Proceedings of the 12th International Conference on the European Energy Market-EEM15, Lisbon, Portugal, 19–22 May 2015. [Google Scholar]
- Pleßmann, G.; Erdmann, M.; Hlusiak, M.; Breyer, C. Global energy storage demand for a 100% renewable electricity supply. Energy Procedia 2014, 46, 22–31. [Google Scholar] [CrossRef]
- Kalghatgi, G. Is it really the end of internal combustion engines and petroleum in transport? Appl. Energy 2018, 225, 965–974. [Google Scholar] [CrossRef]
- Schemme, S.; Samsun, R.C.; Peters, R.; Stolten, D. Power-to-fuel as a key to sustainable transport systems—An analysis of diesel fuels produced from CO2 and renewable electricity. Fuel 2017, 205, 198–221. [Google Scholar] [CrossRef]
- Fuchs, G.; Lunz, B.; Leuthold, M.; Sauer, D.U. Technology Overview on Electricity Storage Overview on the Potential and on the Deployment Perspectives of Electricity Storage Technologies; Smart Energy for Europe Platform GmbH (SEFEP): Berlin, Germany, 2012. [Google Scholar]
- Niaz, S.; Manzoor, T.; Pandith, A.H. Hydrogen storage: Materials, methods and perspectives. Renew. Sustain. Energy Rev. 2015, 50, 457–469. [Google Scholar] [CrossRef]
- Fuel Cell Technologies Office. Multi-Year Research, Development, and Demonstration Plan. Available online: https://www.energy.gov/eere/fuelcells/downloads/hydrogen-consortium-overview-part-2-3-electrolysis-webinar (accessed on 9 May 2019).
- EnergyLab 2.0 Project Website. Available online: https://www.elab2.kit.edu/ (accessed on 25 March 2019).
- Gill, S.S.; Tsolakis, A.; Dearn, K.D.; Rodríguez-Fernández, J. Combustion characteristics and emissions of Fischer-Tropsch diesel fuels in IC engines. Prog. Energy Combust. 2011, 37, 503–523. [Google Scholar] [CrossRef]
- Tschann, P. Emission and Performance Studies of Alternative Fuels. Master’s Thesis, Graz University of Technology, Graz, Austria, 2009. [Google Scholar]
- Abu-Jrai, A.; Tsolakis, A.; Theinnoi, K.; Cracknell, R.; Megaritis, A.; Wyszynski, M.L.; Golunski, S.E. Effect of gas-to-liquid diesel fuels on combustion characteristics, engine emissions, and exhaust gas fuel reforming. Comparative study. Energy Fuel 2006, 20, 2377–2384. [Google Scholar] [CrossRef]
- Fischer-Tropsch Synthesis: Catalysts and Chemistry; Van de Loosdrecht, J., Botes, F.G., Ciobica, I.M., Ferreira, A., Gibson, P., Moodley, D.J., Saib, A.M., Visagie, J.L., Weststrate, C.J., Niemantsverdriet, J.W., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; ISBN 9780080965291. [Google Scholar]
- Albrecht, F.G.; König, D.H.; Baucks, N.; Dietrich, R.-U. A standardized methodology for the techno-economic evaluation of alternative fuels—A case study. Fuel 2017, 194, 511–526. [Google Scholar] [CrossRef]
- Siegemund, S.; Trommler, M.; Kolb, O.; Zinnecker, V.; Schmidt, P.; Weindorf, W.; Zittel, W.; Raksha, T.; Zerhusen, J. The Potential of Electricity-Based Fuels for Low-Emission Transport in the EU an Expertise by LBST and Dena; Deutsche Energie-Agentur GmbH (dena): Berlin, Germany, 2017. [Google Scholar]
- LeViness, S.; Deshmukh, S.R.; Richard, L.A.; Robota, H.J. Velocys Fischer-Tropsch synthesis technology—New advances on state-of-the-art. Top. Catal. 2014, 57, 518–525. [Google Scholar] [CrossRef]
- Kalz, K.F.; Kraehnert, R.; Dvoyashkin, M.; Dittmeyer, R.; Gläser, R.; Krewer, U.; Reuter, K.; Grunwaldt, J.D. Future challenges in heterogeneous catalysis: Understanding catalysts under dynamic reaction conditions. ChemCatChem 2017, 9, 17–29. [Google Scholar] [CrossRef]
- Venvik, H.J.; Yang, J. Catalysis in microstructured reactors: Short review on small-scale syngas production and further conversion into methanol, DME and Fischer-Tropsch products. Catal. Today 2017, 285, 135–146. [Google Scholar] [CrossRef]
- Steynberg, A.P.; Deshmukh, S.R.; Robota, H.J. Fischer-Tropsch catalyst deactivation in commercial microchannel reactor operation. Catal. Today 2018, 299, 10–13. [Google Scholar] [CrossRef]
- Van Sint Annaland, M. Editorial overview: Process intensification in reaction engineering: Intensified efforts to boost intensification. Curr. Opin. Chem. Eng. 2017, 17, ii–iii. [Google Scholar] [CrossRef]
- Kolb, G. Review: Microstructured reactors for distributed and renewable production of fuels and electrical energy. Chem. Eng. Process. 2013, 65, 1–44. [Google Scholar] [CrossRef]
- Tonkovich, A.; Mazanec, T.; Jarosch, K. Improved Fischer-Tropsch catalysts. Focus Catal. 2004, 2004, 7. [Google Scholar] [CrossRef]
- Delparish, A.; Avci, A.K. Intensified catalytic reactors for Fischer-Tropsch synthesis and for reforming of renewable fuels to hydrogen and synthesis gas. Fuel Process. Technol. 2016, 151, 72–100. [Google Scholar] [CrossRef]
- Cao, C.; Hu, J.; Li, S.; Wilcox, W.; Wang, Y. Intensified Fischer-Tropsch synthesis process with microchannel catalytic reactors. Catal. Today 2009, 140, 149–156. [Google Scholar] [CrossRef]
- Kshetrimayum, K.S.; Jung, I.; Na, J.; Park, S.; Lee, Y.; Park, S.; Lee, C.J.; Han, C. CFD simulation of microchannel reactor block for Fischer-Tropsch synthesis: Effect of coolant type and wall boiling condition on reactor temperature. Ind. Eng. Chem. 2016, 55, 543–554. [Google Scholar] [CrossRef]
- Almeida, L.C.; Sanz, O.; D’Olhaberriague, J.; Yunes, S.; Montes, M. Microchannel reactor for Fischer-Tropsch synthesis: Adaptation of a commercial unit for testing microchannel blocks. Fuel 2013, 110, 171–177. [Google Scholar] [CrossRef]
- Arzamendi, G.; Diéguez, P.M.; Montes, M.; Odriozola, J.A.; Sousa-Aguiar, E.F.; Gandía, L.M. Computational fluid dynamics study of heat transfer in a microchannel reactor for low-temperature Fischer-Tropsch synthesis. Chem. Eng. J. 2010, 160, 915–922. [Google Scholar] [CrossRef]
- Dittmeyer, R.; Boeltken, T.; Piermartini, P.; Selinsek, M.; Loewert, M.; Dallmann, F.; Kreuder, H.; Cholewa, M.; Wunsch, A.; Belimov, M.; et al. Micro and micro membrane reactors for advanced applications in chemical energy conversion. Curr. Opin. Chem. Eng. 2017, 17, 108–125. [Google Scholar] [CrossRef]
- LeViness, S. Opportunities for modular GTL in North America. In Proceedings of the Energy Frontiers International, Gas-to-Market & Energy Conversion Forum, Houston, TX, USA, 22 October 2012. [Google Scholar]
- LeViness, S. Velocys Fischer-Tropsch synthesis technology comparison to conventional FT technologies. In Proceedings of the AIChE 2013 Spring Meeting, San Francisco, CA, USA, 3–8 November 2013; AIChE: San Antonio, TX, USA, 2013. [Google Scholar]
- Zhang, J.; Xu, G. Scale-up of bubbling fluidized beds with continuous particle flow based on particle-residence-time distribution. Particuology 2015, 19, 155–163. [Google Scholar] [CrossRef]
- Sievers, D.A.; Kuhn, E.M.; Stickel, J.J.; Tucker, M.P.; Wolfrum, E.J. Online residence time distribution measurement of thermochemical biomass pretreatment reactors. Chem. Eng. Sci. 2016, 140, 330–336. [Google Scholar] [CrossRef]
- Eilers, H.; González, M.I.; Schaub, G. Lab-scale experimental studies of Fischer-Tropsch kinetics in a three-phase slurry reactor under transient reaction conditions. Catal. Today 2016, 275, 164–171. [Google Scholar] [CrossRef]
- González, M.I.; Schaub, G. Fischer-Tropsch synthesis with H2/CO2-catalyst behavior under transient conditions. Chem. Ing. Tech. 2015, 87, 848–854. [Google Scholar] [CrossRef]
- Schlosser, E.; Wolfrum, J.; Hildebrandt, L.; Seifert, H.; Oser, B.; Ebert, V. Diode laser based in situ detection of alkali atoms: Development of a new method for determination of residence-time distribution in combustion plants. Appl. Phys. B 2002, 75, 237–247. [Google Scholar] [CrossRef]
- Levenspiel, O. Tracer Technology; Springer: New York, NY, USA, 2012; ISBN 978-1-4419-8073-1. [Google Scholar]
- Van Steen, E.; Claeys, M.; Möller, K.P.; Nabaho, D. Comparing a cobalt-based catalyst with iron-based catalysts for the Fischer-Tropsch XTL-process operating at high conversion. Appl. Catal. A Gen. 2018, 549, 51–59. [Google Scholar] [CrossRef]
- Fischer-Tropsch Technology; Steynberg, A., Dry, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; ISBN 9780444513540. [Google Scholar]
- VDI Heat Atlas, 2nd ed.; VDI Gesellschaft Verfahrenstechnik und Ingenieurwesen, Ed.; Springer: Berlin/Heidelberg, Germany, 2010; ISBN 9783540799993. [Google Scholar]
- Kreitz, B.; Wehinger, G.D.; Turek, T. Dynamic simulation of the CO2 methanation in a micro-structured fixed-bed reactor. Chem. Eng. Sci. 2019, 195, 541–552. [Google Scholar] [CrossRef]
- Adesina, A.A.; Silveston, P.L.; Hudgins, R.R. A comparison of forced feed cycling of the Fischer-Tropsch synthesis over iron and cobalt catalysts. In Studies in Surface Science and Catalysis; Kaliaguine, S., Mahay, A., Eds.; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Silveston, P.; Hudgins, R.R.; Renken, A. Periodic operation of catalytic reactors-introduction and overview. Catal. Today 1995, 25, 91–112. [Google Scholar] [CrossRef]
- Topsøe, H. Developments in operando studies and in situ characterization of heterogeneous catalysts. J. Catal. 2003, 216, 155–164. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, D.; Xu, X.; Sheng, Y.; Xu, J.; Han, Y.F. Application of operando spectroscopy on catalytic reactions. Curr. Opin. Chem. Eng. 2016, 12, 1–7. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Ford, M.E.; Gregory, D.; Hu, R.; Keturakis, C.J.; Lwin, S.; Tang, Y.; Yang, Z.; Zhu, M.; Bañares, M.A.; et al. A decade+ of operando spectroscopy studies. Catal. Today 2017, 283, 27–53. [Google Scholar] [CrossRef]
- Rochet, A.; Moizan, V.; Pichon, C.; Diehl, F.; Berliet, A.; Briois, V. In situ and operando structural characterisation of a Fischer-Tropsch supported cobalt catalyst. Catal. Today 2011, 171, 186–191. [Google Scholar] [CrossRef]
- Jacobs, G.; Ma, W.; Gao, P.; Todic, B.; Bhatelia, T.; Bukur, D.B.; Davis, B.H. The application of synchrotron methods in characterizing iron and cobalt Fischer-Tropsch synthesis catalysts. Catal. Today 2013, 214, 100–139. [Google Scholar] [CrossRef]
- Karlsruhe Institute of Technology (KIT). PowerFuel—Fuels for Climate-Neutral Airplanes. Available online: https://www.kit.edu/kit/english/pi_2018_165_fuels-for-climate-neutral-airplanes.php (accessed on 9 May 2019).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System Component | Volume (L) | Temperature (°C) |
|---|---|---|
| Reactor | 0.15 | 220–250 |
| HT/µHE CT | 4.8 25 | 170 10 |
| Mode | Setup | Total Gas Flow (L min−1) | H2 Content (vol.%) | N2 Content (vol.%) | CO Content (vol.%) | CO2 Content (vol.%) | Temperature (°C) | Pressure (MPa) | H2/CO (-) |
|---|---|---|---|---|---|---|---|---|---|
| Non-reactive | CO2 on/off | 20 | 95 | 5/0 | 0 | 0/5 | 225 | 3 | - |
| Reactive | H2/CO high | 17 | 50 | 25 | 25 | 0 | 238 | 3 | 1.95 |
| Reactive | H2/CO low | 17 | 35 | 40 | 25 | 0 | 237 | 3 | 1.38 |
| Species/Property | Concentration |
|---|---|
| Methanol | 14.19 ± 4.14 g L−1 |
| Ethanol | 4.64 ± 1.44 g L−1 |
| Propanol | 1.21 ± 0.32 g L−1 |
| Butanol | 0.79 ± 0.27 g L−1 |
| Pentanol | 0.66 ± 0.19 g L−1 |
| Hexanol | 0.23 ± 0.06 g L−1 |
| Heptanol | 0.07 ± 0.02 g L−1 |
| Total concentration | 21.78 ± 6.04 g L−1 |
| Uncalibrated peaks | 7.41 ± 2.10 % |
| Property | Content/Value |
|---|---|
| Av. alkane content (oil) | 78.18 ± 3.21 wt.% |
| Av. alkene content (oil) | 14.14 ± 4.03 wt.% |
| Av. iso-alkane and alcohol content (oil) | 7.69 ± 1.78 wt.% |
| Av. carbon chain length (oil) | 10.71 ± 0.79 atoms |
| Av. probability of chain growth (α) | 88.44 ± 1.53% |
| Property | QS | CT |
|---|---|---|
| Alkane content (oil) | 80.22 wt.% | 75.77 wt.% |
| Alkene content (oil) | 9.50 wt.% | 17.70 wt.% |
| Iso-alkane and alcohol content (oil) | 10.28 wt.% | 6.53 wt.% |
| Carbon chain length (oil) | 11.47 atoms | 9.56 atoms |
| Setup | H2 Conversion (vol.%) | CO Conversion (vol.%) | SC1 (mol.%) | SC5+ (mol.%) | Alkane Content (wt.%) | Alkene Content (wt.%) | Iso-Alkane + Alcohol Content (wt.%) | Av. Carbon Chain Length (-) |
|---|---|---|---|---|---|---|---|---|
| H2/CO high | 68.43 | 62.71 | 12.68 | 78.38 | 83.74 | 10.57 | 5.68 | 11.99 |
| H2/CO low | 63.20 | 41.09 | 9.53 | 82.74 | 78.02 | 14.31 | 7.67 | 12.37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loewert, M.; Pfeifer, P. Dynamically Operated Fischer-Tropsch Synthesis in PtL-Part 1: System Response on Intermittent Feed. ChemEngineering 2020, 4, 21. https://doi.org/10.3390/chemengineering4020021
Loewert M, Pfeifer P. Dynamically Operated Fischer-Tropsch Synthesis in PtL-Part 1: System Response on Intermittent Feed. ChemEngineering. 2020; 4(2):21. https://doi.org/10.3390/chemengineering4020021
Chicago/Turabian StyleLoewert, Marcel, and Peter Pfeifer. 2020. "Dynamically Operated Fischer-Tropsch Synthesis in PtL-Part 1: System Response on Intermittent Feed" ChemEngineering 4, no. 2: 21. https://doi.org/10.3390/chemengineering4020021
APA StyleLoewert, M., & Pfeifer, P. (2020). Dynamically Operated Fischer-Tropsch Synthesis in PtL-Part 1: System Response on Intermittent Feed. ChemEngineering, 4(2), 21. https://doi.org/10.3390/chemengineering4020021