
Dichloroacetyl Amides of 3,5-Bis(benzylidene)-4-piperidones Displaying Greater Toxicity to Neoplasms than to Non-Malignant Cells

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Experimental Methods
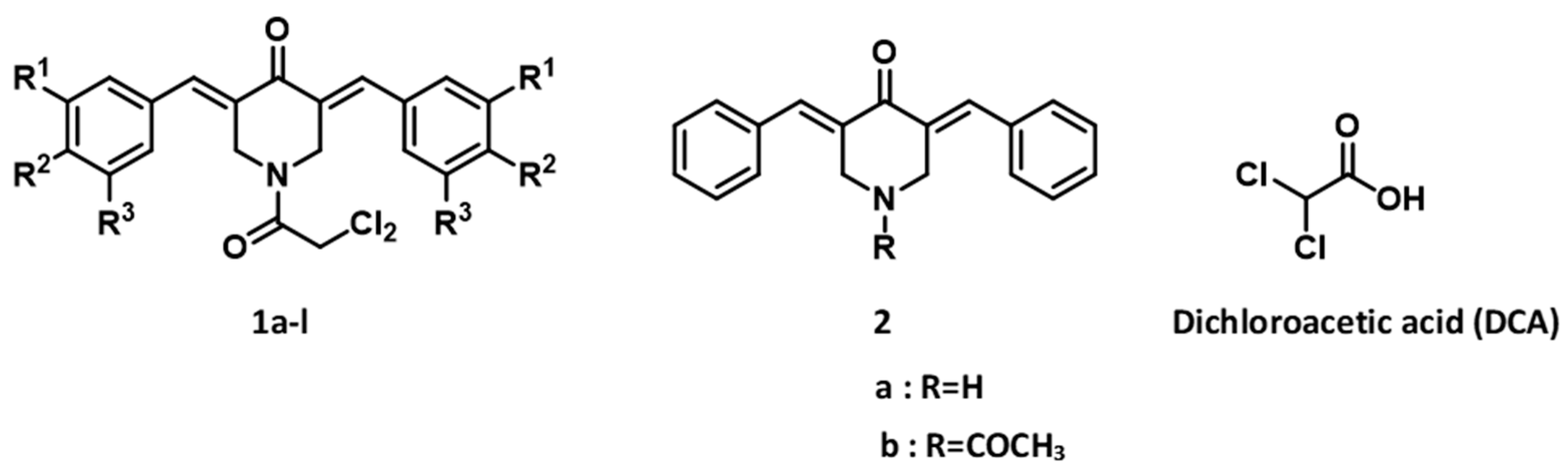
2.1. Synthesis of Compounds
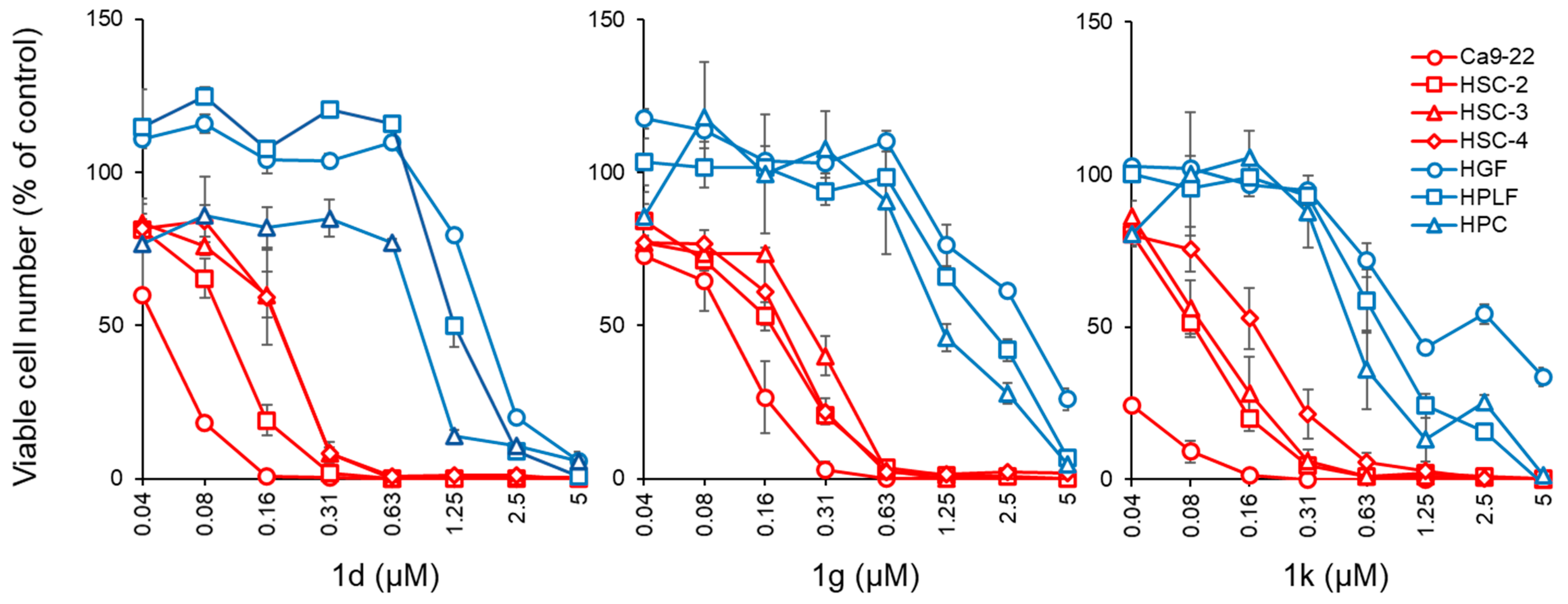
2.2. Cytotoxicity Assays
2.3. Calculation of Selectivity Index (SI)
2.4. Calculation of Potency-Selectivity Expression (PSE)
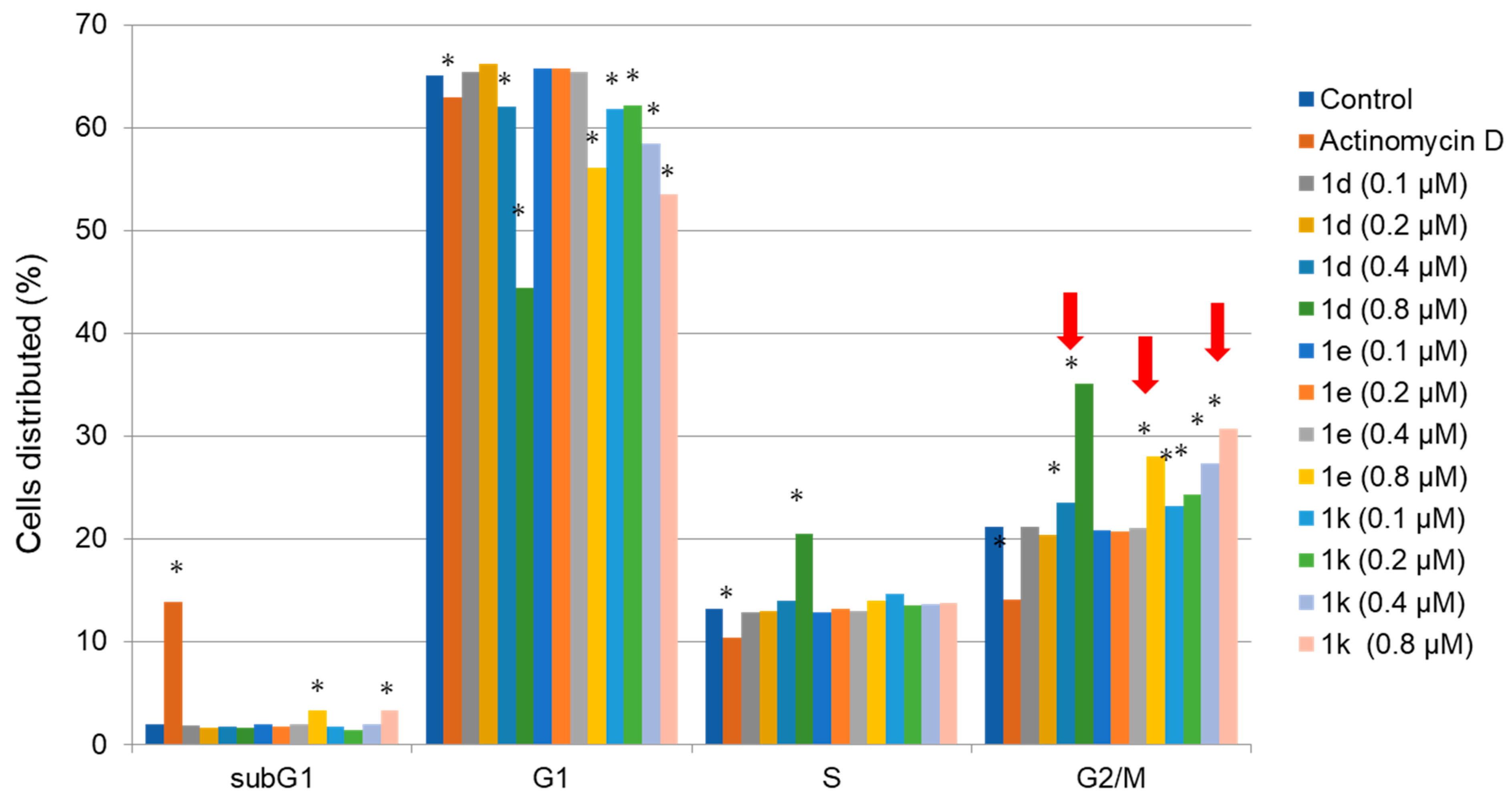
2.5. Cell Cycle Assay
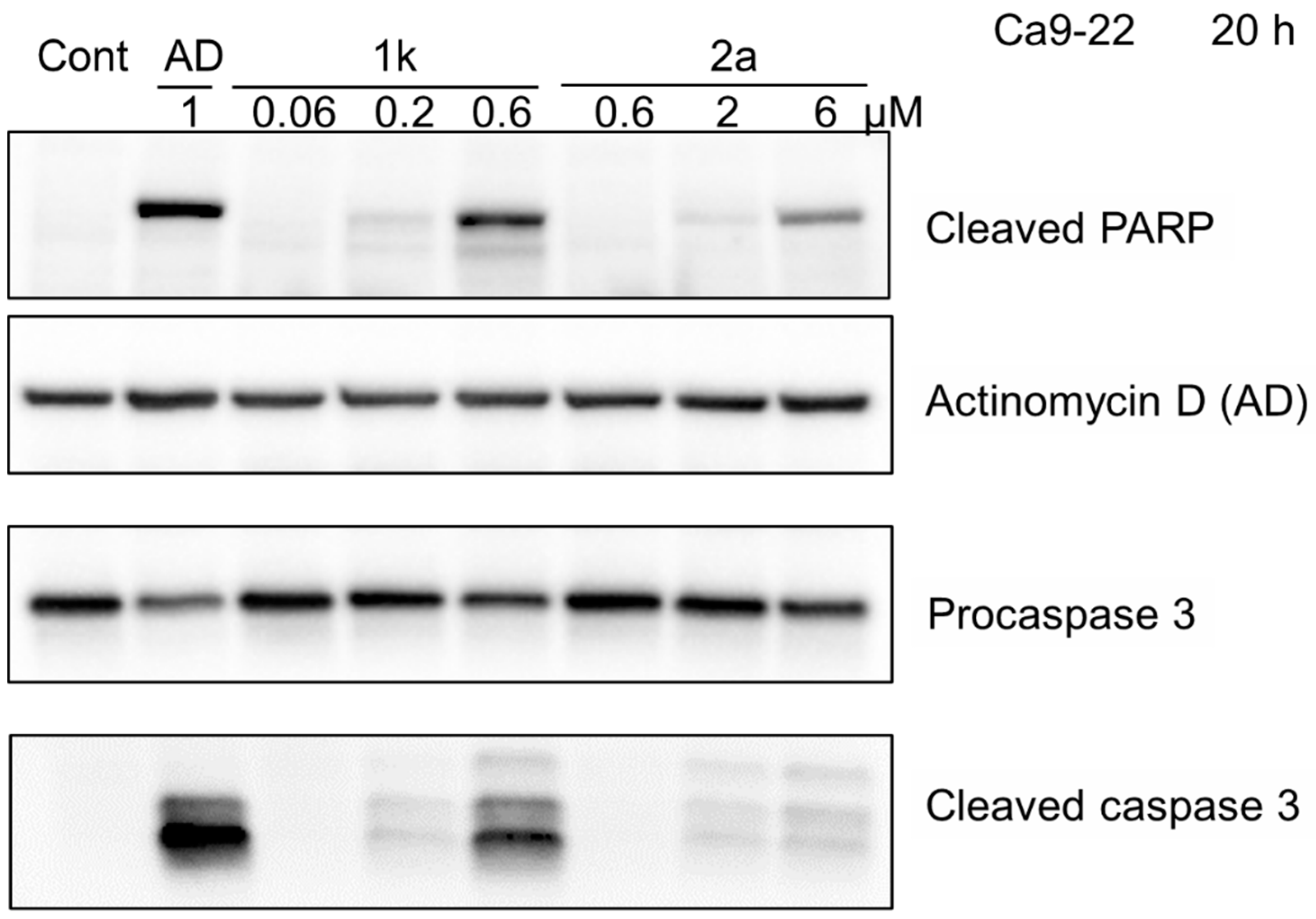
2.6. PARP and Procaspase 3 Cleavage
2.7. Statistical Treatment
3. Results
4. Discussion
- Linear graphs were made between the average CC50 values of 1a–l and the σ values, the π constants, and subsequently with the MR figures.
- Linear graphs were prepared between the average SI values of 1a–l and the σ, π, and MR constants.
- Linear graphs were made between the PSE values of 1a–l and the σ, π, and MR constants.
- Stages 1–3 were repeated, except semilogarithmic were made, not linear plots.
- Stages 1–3 were repeated, except correlations were sought with the data for 1a–k, i.e., the outlier 1l was removed from consideration.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ivasiv, V.; Albertini, C.; Goncalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular hybridization as a tool for designing multitarget candidates for complex diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Hossain, H.; Das, S.; Das, U.; Doroudi, A.; Zhu, J.; Dimmock, J.R. Novel hybrid molecules of 3,5-bis(benzylidene)-4-piperidones and dichloroacetic acid which demonstrate potent tumour-selective cytotoxicity. Bioorganic Med. Chem. Lett. 2020, 30, 126878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.M.S.; Herrmann, L.; Tsogoeva, S.B. Structural hybridization as a facile approach to new drug candidates. Bioorganic Med. Chem. Lett. 2020, 30, 127514. [Google Scholar] [CrossRef] [PubMed]
- Hur, H.; Xuan, Y.; Kim, Y.B.; Lee, G.; Shim, W.; Yun, J.; Ham, I.H.; Han, S.U. Expression of pyruvate dehydrogenase kinase -1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 2013, 42, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Hu, X.; Zhang, W.; Yao, H.; Tam, K.Y. Development of pyruvate dehydrogenase kinase inhibitors in medicinal chemistry with particular references as anticancer agents. Drug Discov. Today 2015, 20, 1112–1119. [Google Scholar] [CrossRef]
- Wei, G.; Sun, J.; Luan, W.; Hou, Z.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Natural product albiziabioside a conjugated with pyruvate dehydrogenase kinase inhibitor dichloroacetate to induce apoptosis-ferroptosis-M2-TAMs polarization for combined cancer therapy. J. Med. Chem. 2019, 62, 8760–8772. [Google Scholar] [CrossRef]
- Yang, C.; Wu, T.; Qin, Y.; Qi, Y.; Sun, Y.; Kong, M.; Jiang, X.; Qin, X.; Shen, Y.; Zhang, Z. A facile doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery. Int. J. Nanomed. 2018, 13, 1281–1293. [Google Scholar] [CrossRef] [Green Version]
- Ghinet, A.; Thuru, X.; Floquet, E.; Dubois, J.; Farce, A.; Rigo, B. Enhanced antitumor potential induced by chloroacetate-loaded benzophenones acting as fused tubulin-pyruvate dehydrogenase kinase 1 (PDHK1) ligands. Bioorganic Chem. 2020, 96, 103643. [Google Scholar] [CrossRef]
- Bonner, M.Y.; Karlsson, I.; Rodolfo, M.; Arnold, R.S.; Vergani, E.; Arbiser, J.L. Honokiol bis-dichloroacetate (Honokiol DCA) demonstrates activity in vemurafenib-resistant melanoma In Vivo. Oncotarget 2016, 7, 12857–12868. [Google Scholar] [CrossRef]
- Trapella, C.; Voltan, R.; Melloni, E.; Tisato, V.; Celeghini, C.; Bianco, S.; Fantinati, A.; Salvadori, S.; Guerrini, R.; Secchiero, P.; et al. Design, synthesis, and biological characterization of novel mitochondria targeted dichloroacetate-loaded compounds with antileukemic activity. J. Med. Chem. 2016, 59, 147–156. [Google Scholar] [CrossRef]
- Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: A mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem. Biol. 2014, 9, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Lippard, S.J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. USA 2009, 106, 22199–22204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Dong, X.; Shi, Q.; Chen, J.; Su, W. Improving the anticancer activity of platinum(IV) prodrugs using a dual-targeting strategy with a dichloroacetate axial ligand. RSC Adv. 2019, 9, 22240–22247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fereidoonnezhad, M.; Tabaei, S.M.H.; Sakhteman, A.; Seradj, H.; Faghih, Z.; Faghih, Z.; Mojaddami, A.; Sadeghian, B.; Rezaei, Z. Design, synthesis, molecular docking, biological evaluations and QSAR studies of novel dichloroacetate analogues as anticancer agent. J. Mol. Struct. 2020, 1221, 128689. [Google Scholar] [CrossRef]
- Sanchez-Martin, C.; Menon, D.; Moroni, E.; Ferraro, M.; Masgras, I.; Elsey, J.; Arbiser, J.L.; Colombo, G.; Rasola, A. Honokiol bis-dichloroacetate is a selective allosteric inhibitor of the mitochondrial chaperone TRAP1. Antioxid. Redox Signal. 2021, 34, 505–516. [Google Scholar] [CrossRef]
- Hossain, M.; Das, U.; Dimmock, J.R. Recent advances in α,β-unsaturated carbonyl compounds as mitochondrial toxins. Eur. J. Med. Chem. 2019, 183, 111687. [Google Scholar] [CrossRef]
- Kantoh, K.; Ono, M.; Nakamura, Y.; Nakamura, Y.; Hashimoto, K.; Sakagami, H.; Wakabayashi, H. Hormetic and anti-radiation effects of tropolone-related compounds. In Vivo 2010, 24, 843–851. [Google Scholar]
- Sakagami, H.; Shimada, C.; Kanda, Y.; Amano, O.; Sugimoto, M.; Ota, S.; Soga, T.; Tomita, M.; Sato, A.; Tanuma, S.-I.; et al. Effects of 3-styrylchromones on metabolic profiles and cell death in oral squamous cell carcinoma cells. Toxicol. Rep. 2015, 2, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Iijima, Y.; Bandow, K.; Sano, M.; Hino, S.; Kaneko, T.; Horie, N.; Sakagami, H. In Vitro assessment of antitumor potential and combination effect of classical and molecular-targeted anticancer drugs. Anticancer Res. 2019, 39, 6673–6684. [Google Scholar] [CrossRef] [Green Version]
- Yamali, C.; Sakagami, H.; Uesawa, Y.; Kurosaki, K.; Satoh, K.; Masuda, Y.; Yokose, S.; Ece, A.; Bua, S.; Angeli, A.; et al. Comprehensive study on potent and selective carbonic anhydrase inhibitors: Synthesis, bioactivities and molecular modelling studies of 4-(3-(2-arylidenehydrazine-1-carbonyl)-5-(thiophen-2-yl)-1H-pyrazole-1-yl) benzenesulfonamides. Eur. J. Med. Chem. 2021, 217, 113351. [Google Scholar] [CrossRef]
- Taghavi, N.; Yazdi, I. Prognostic factors of survival rate in oral squamous cell carcinoma: Clinical, histologic, genetic and molecular concepts. Arch. Iran. Med. 2015, 18, 314–319. [Google Scholar] [PubMed]
- Hansch, C.; Leo, A.J. Substituent Constants for Correlation Analysis in Chemistry and Biology; John Wiley and Sons: New York, NY, USA, 1979; pp. 49–50. [Google Scholar]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst.) 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Aryl Group | Ca9-22 CC50 (µM) | SI a | HSC-2 CC50 (µM) | SI a | HSC-3 CC50 (µM) | SI a | HSC-4 CC50 (µM) | SI a | Average CC50 (µM) | SI a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | H | 0.42 ± 0.04 | 9.64 | 0.67 ± 0.06 | 6.05 | 1.29 ± 0.07 | 3.14 | 0.59 ± 0.12 | 6.86 | 0.74 | 6.42 |
| 1b | 4-F | 0.11 ± 0.01 | 19.8 | 0.25 ± 0.02 | 8.72 | 0.44 ± 0.02 | 4.96 | 0.33 ± 0.07 | 6.61 | 0.28 | 10.0 |
| 1c | 3,4-F2 | 0.09 ± 0.02 | 20.0 | 0.20 ± 0.02 | 9.00 | 0.35 ± 0.02 | 5.14 | 0.20 ± 0.01 | 9.00 | 0.21 | 10.8 |
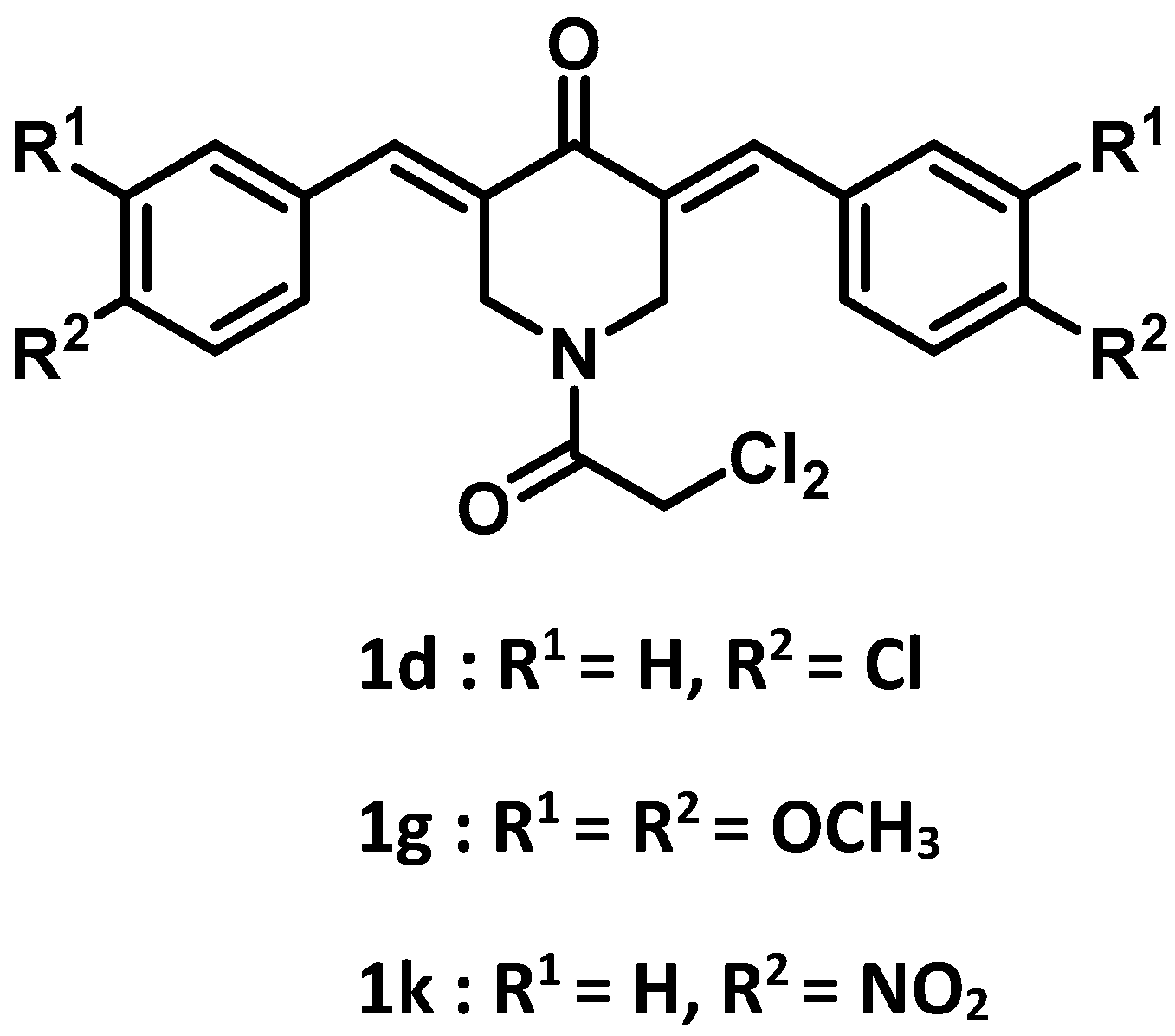
| 1d | 4-Cl | 0.05 ± 0.00 | 28.8 | 0.10 ± 0.01 | 14.4 | 0.19 ± 0.02 | 7.58 | 0.18 ± 0.04 | 8.00 | 0.13 | 14.7 |
| 1e | 3,4-Cl2 | 0.04 ± 0.00 | 19.5 | 0.08 ± 0.01 | 9.75 | 0.13 ± 0.06 | 6.00 | 0.23 ± 0.01 | 3.39 | 0.12 | 9.66 |
| 1f | 4-OCH3 | 0.39 ± 0.02 | 15.4 | 0.52 ± 0.07 | 11.5 | 1.00 ± 0.16 | 5.99 | 1.26 ± 0.27 | 4.75 | 0.79 | 9.41 |
| 1g | 3,4-(OCH3)2 | 0.11 ± 0.02 | 36.8 | 0.17 ± 0.02 | 23.8 | 0.28 ± 0.03 | 14.5 | 0.20 ± 0.04 | 20.3 | 0.19 | 23.9 |
| 1h | 3,4,5-(OCH3)3 | 0.72 ± 0.15 | 11.9 | 0.14 ± 0.01 | 61.3 | 1.37 ± 0.22 | 6.26 | 0.99 ± 0.39 | 8.67 | 0.80 | 22.0 |
| 1i | 3,4-OCH2O | 0.17 ± 0.06 | 25.1 | 0.36 ± 0.01 | 11.9 | 0.74 ± 0.13 | 5.77 | 0.65 ± 0.17 | 6.57 | 0.48 | 12.3 |
| 1j | 4-CH3 | 0.15 ± 0.03 | 32.1 | 0.26 ± 0.01 | 18.5 | 0.46 ± 0.04 | 10.5 | 0.45 ± 0.04 | 10.7 | 0.33 | 18.0 |
| 1k | 4-NO2 | 0.02 ± 0.01 | 72.0 | 0.08 ± 0.01 | 18.0 | 0.10 ± 0.03 | 14.4 | 0.18 ± 0.04 | 8.00 | 0.10 | 28.1 |
| 1l | 4-N(CH3)2 | 13.6 ± 2.70 | 10.3 | 14.9 ± 4.40 | 9.40 | 45.5 ± 7.60 | 3.08 | 19.6 ± 6.30 | 7.14 | 23.4 | 7.48 |
| 2a | - | 0.19 ± 0.03 | 61.6 | 0.39 ± 0.01 | 30.0 | 0.80 ± 0.07 | 14.6 | 0.79 ± 0.22 | 14.8 | 0.54 | 30.3 |
| 2b | - | 0.64 ± 0.01 | 17.3 | 1.23 ± 0.07 | 9.02 | 2.25 ± 0.08 | 4.93 | 1.57 ± 0.12 | 7.07 | 1.42 | 9.58 |
| SDA b | >200 | - | >200 | - | >200 | - | >200 | - | >200 | - | |
| 5-FU c | 24.5 ± 12.3 | >40.7 | 30.5 ± 7.1 | >32.7 | 61.3 ± 9.8 | >16.3 | 7.58 ± 0.5 | >131 | 31.0 | >55.2 | |
| DXR d | 0.43 ± 0.04 | >22.2 | 0.20 ± 0.02 | >47.8 | 0.26 ± 0.21 | >36.7 | 0.12 ± 0.00 | >79.6 | 0.25 | >46.6 |
| Compound | Aryl Group | CC50 (µM) | PSE a | |||
|---|---|---|---|---|---|---|
| HGF | HPLF | HPC | Average | |||
| 1a | H | 5.90 ± 0.10 | 3.37 ± 0.42 | 2.88 ± 0.08 | 4.05 | 868 |
| 1b | 4-F | 2.97 ± 0.46 | 2.26 ± 0.11 | 1.30 ± 0.16 | 2.18 | 3571 |
| 1c | 3,4-F2 | 2.21 ± 0.11 | 1.91 ± 0.08 | 1.28 ± 0.37 | 1.80 | 5143 |
| 1d | 4-Cl | 2.13 ± 0.12 | 1.29 ± 0.14 | 0.90 ± 0.02 | 1.44 | 11,308 |
| 1e | 3,4-Cl2 | 1.04 ± 0.13 | 0.82 ± 0.10 | 0.49 ± 0.00 | 0.78 | 8050 |
| 1f | 4-OCH3 | 9.03 ± 0.76 | 5.33 ± 0.32 | 3.62 ± 0.07 | 5.99 | 1191 |
| 1g | 3,4-(OCH3)2 | 8.13 ± 2.47 | 2.83 ± 1.53 | 1.19 ± 0.09 | 4.05 | 12,579 |
| 1h | 3,4,5-(OCH3)3 | 4.70 ± 1.60 | 17.0 ± 5.10 | 4.00 ± 0.60 | 8.58 | 2750 |
| 1i | 3,4-OCH2O | 5.67 ± 0.31 | 4.37 ± 0.85 | 2.78 ± 0.14 | 4.27 | 2563 |
| 1j | 4-CH3 | 5.53 ± 0.31 | 5.00 ± 0.10 | 3.93 ± 0.65 | 4.82 | 5455 |
| 1k | 4-NO2 | 3.01 ± 0.33 | 0.76 ± 0.13 | 0.56 ± 0.08 | 1.44 | 28,100 |
| 1l | 4-N(CH3)2 | 111 ± 27.0 | 188 ± 21.0 | 122 ± 69.0 | 140 | 32.0 |
| 2a | H | 21.7 ± 6.00 | 9.10 ± 2.87 | 4.30 ± 0.72 | 11.7 | 5611 |
| 2b | H | 23.0 ± 1.70 | 5.60 ± 1.13 | 4.83 ± 0.12 | 11.1 | 675 |
| SDA b | >200 | >200 | >200 | >200 | - | |
| 5-FU c | >1000 | >1000 | >987 | >996 | >178 | |
| DXR d | >10 | >10 | >8.65 | >9.55 | >18,640 | |
| Plot | Compounds | Correlations |
|---|---|---|
| Linear | 1a–l | σ (−ve), PSE (+ve) |
| Semilogarithmic | 1a–l | σ (−ve), PSE (+ve) |
| Linear | 1a–k | PSE (+ve) |
| Semilogarithmic | 1a–k | σ (−ve), PSE (+ve) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.; Roayapalley, P.K.; Sakagami, H.; Satoh, K.; Bandow, K.; Das, U.; Dimmock, J.R. Dichloroacetyl Amides of 3,5-Bis(benzylidene)-4-piperidones Displaying Greater Toxicity to Neoplasms than to Non-Malignant Cells. Medicines 2022, 9, 35. https://doi.org/10.3390/medicines9060035
Hossain M, Roayapalley PK, Sakagami H, Satoh K, Bandow K, Das U, Dimmock JR. Dichloroacetyl Amides of 3,5-Bis(benzylidene)-4-piperidones Displaying Greater Toxicity to Neoplasms than to Non-Malignant Cells. Medicines. 2022; 9(6):35. https://doi.org/10.3390/medicines9060035
Chicago/Turabian StyleHossain, Mohammad, Praveen K. Roayapalley, Hiroshi Sakagami, Keitaro Satoh, Kenjiro Bandow, Umashankar Das, and Jonathan R. Dimmock. 2022. "Dichloroacetyl Amides of 3,5-Bis(benzylidene)-4-piperidones Displaying Greater Toxicity to Neoplasms than to Non-Malignant Cells" Medicines 9, no. 6: 35. https://doi.org/10.3390/medicines9060035
APA StyleHossain, M., Roayapalley, P. K., Sakagami, H., Satoh, K., Bandow, K., Das, U., & Dimmock, J. R. (2022). Dichloroacetyl Amides of 3,5-Bis(benzylidene)-4-piperidones Displaying Greater Toxicity to Neoplasms than to Non-Malignant Cells. Medicines, 9(6), 35. https://doi.org/10.3390/medicines9060035