Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan

 ,
,  ,
, 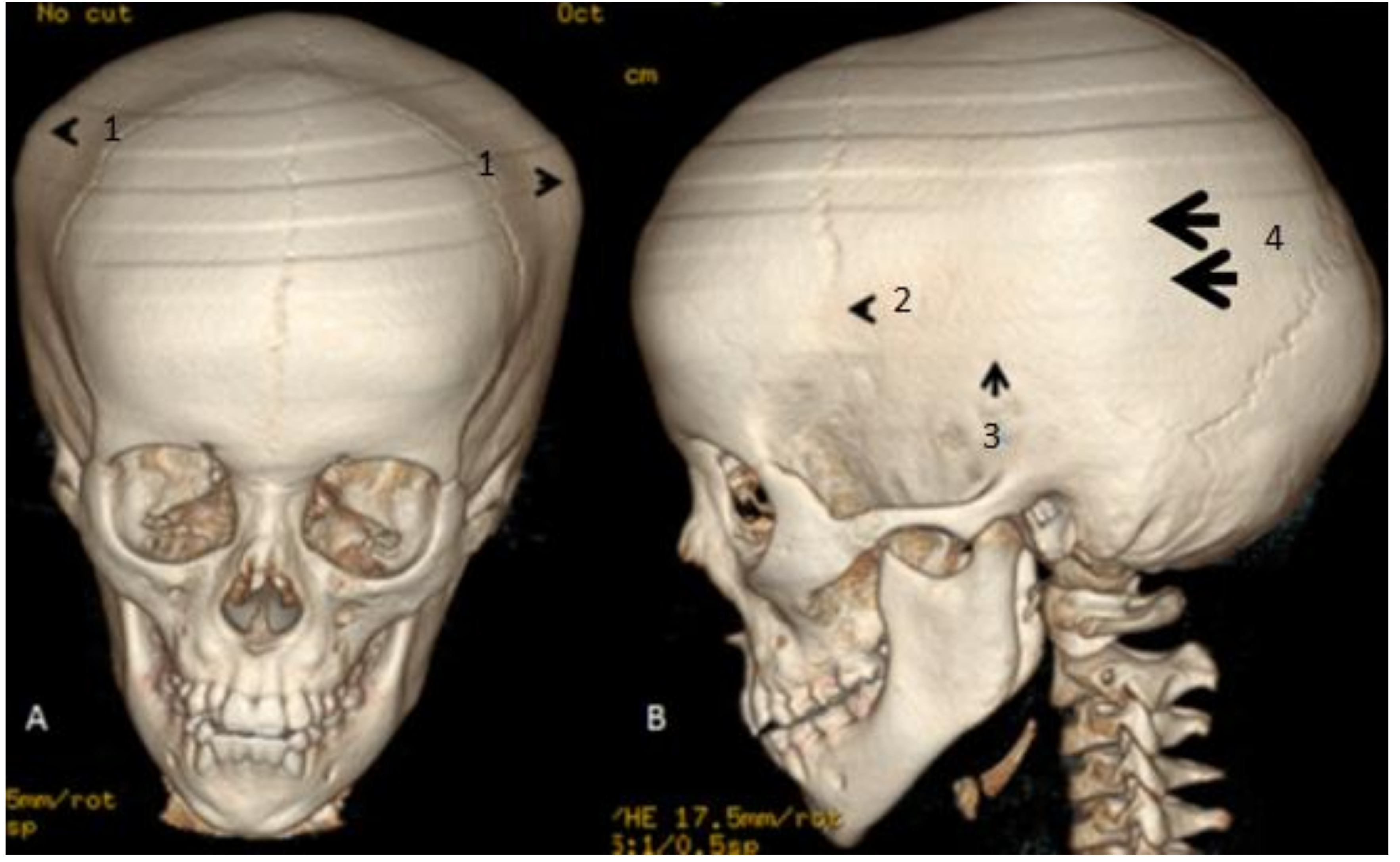{kind=link}
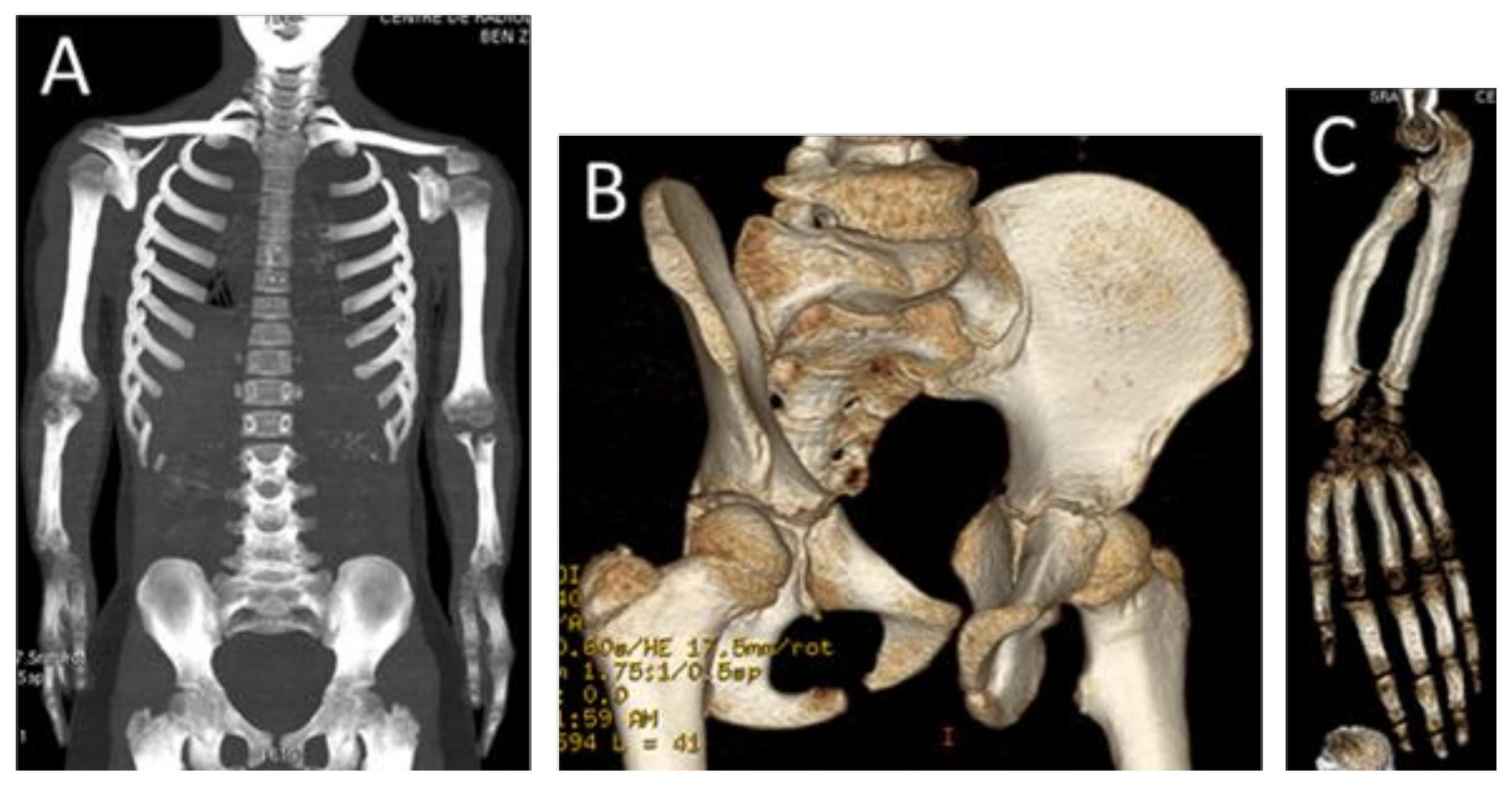{kind=link}
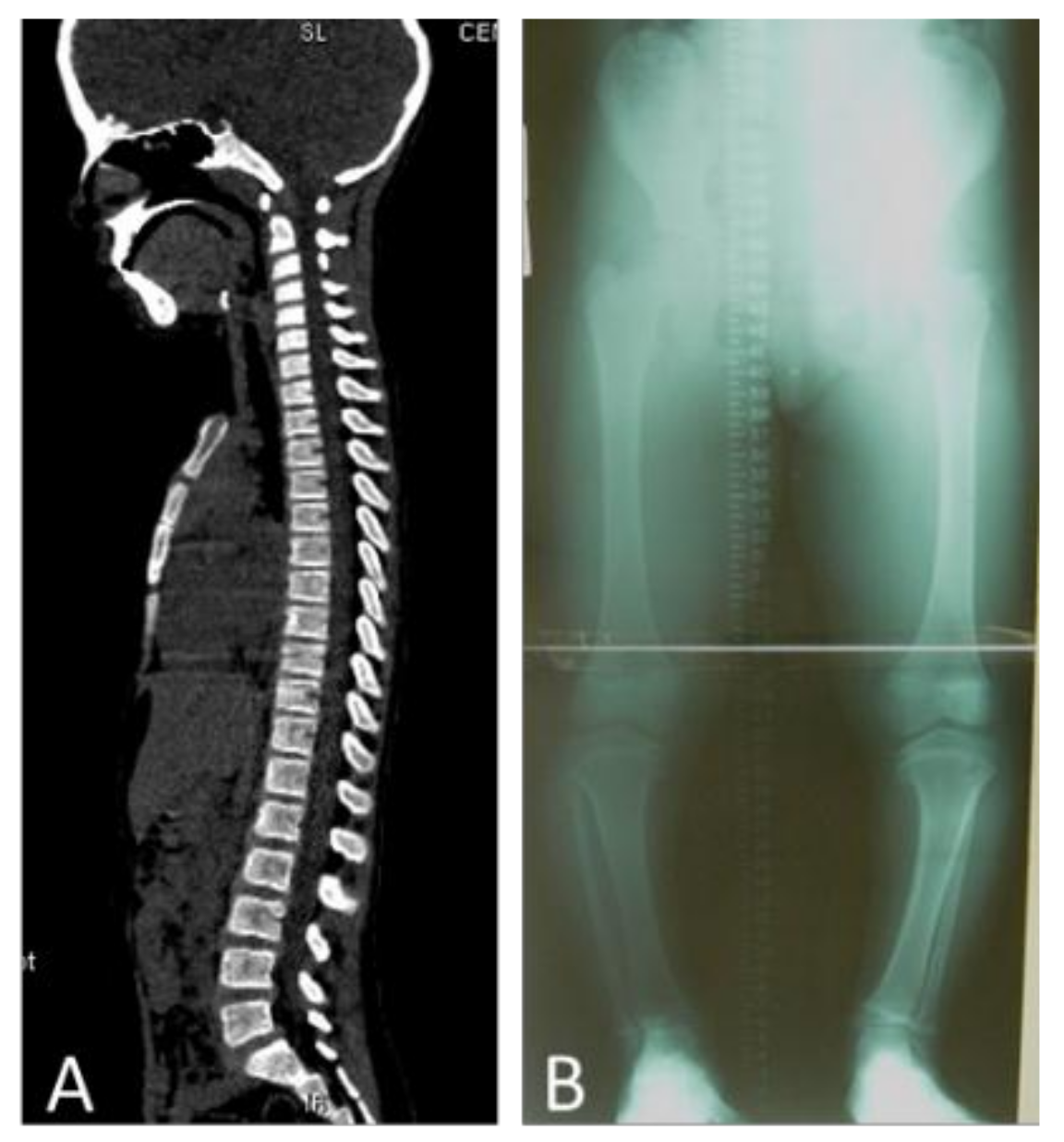{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants and Ethics
2.2. Clinical Examination
2.3. Laboratory Measurements
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leri, A.; Weill, J. Une affection congenitale et symetrique du developpment osseux: la dyschondroosteose. Bull. Mem. Soc. Med. Hop. Paris 1929, 53, 1491–1494. [Google Scholar]
- Binder, G. Short Stature due to SHOX Deficiency: Genotype, Phenotype, and Therapy. Horm. Res. Paediatr. 2011, 75, 81–89. [Google Scholar] [CrossRef]
- Seki, A.; Jinno, T.; Suzuki, E.; Takayama, S.; Ogata, T.; Fukami, M. Skeletal Deformity Associated with SHOX Deficiency. Clin. Pediatr. Endocrinol. 2014, 23, 65–72. [Google Scholar] [CrossRef]
- Corsello, G.; Report, C.; Piccione, M.; Piccione, F.; Giuffrè, M.; De Simone, G.; Peritore, M.; Pierluigi, M. Leri-Weill’s syndrome: Clinical, radiological and genetic investigations in five patients Discondrosteosi di Leri-Weill: Valutazione clinica, radiologica e genetica in cinque pazienti. Rivista Italiana di Pediatria 2006, 32, 55–59. [Google Scholar]
- Maroteaux, P.; Lamy, M. La dyschondrosteose. Sem. Hop. Paris 1959, 35, 3467–3470. [Google Scholar]
- Al Kaissi, A.; Ben Ghachem, M.; Ben Chehida, F.; Hofstaetter, J.G.; Grill, F.; Ganger, R.; Kircher, S.G. Can Multiple Hereditary Exostoses Overlap With Mesomelic Dysplasia? J. Clin. Med. Res. 2016, 8, 605–609. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Langer, L.O. Mesomelic Dwarfism of the Hypoplastic Ulna, Fibula, Mandible Type. Radiology 1967, 89, 654–660. [Google Scholar] [CrossRef]
- Abasq-Thomas, C.; Schmitt, S.; Brenaut, E.; Metz, C.; Chiesa, J.; Misery, L. A Case of Syndromic X-linked Ichthyosis with Léri-Weill Dyschondrosteosis. Acta Derm. Venereol. 2016, 96, 814–815. [Google Scholar] [CrossRef]
- Blaschke, R.J.; Rappold, G. The pseudoautosomal regions, SHOX and disease. Curr. Opin. Genet. Dev. 2006, 16, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Benito-Sanz, S.; Royo, J.L.; Barroso, E.; Paumard-Hernández, B.; Barreda-Bonis, A.C.; Liu, P.; Gracía, R.; Lupski, J.R.; Campos-Barros, Á.; Gómez-Skarmeta, J.L.; et al. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 2012, 49, 442–450. [Google Scholar] [CrossRef]
- Shima, H.; Tanaka, T.; Kamimaki, T.; Dateki, S.; Muroya, K.; Horikawa, R.; Kanno, J.; Adachi, M.; Naiki, Y.; Tanaka, H.; et al. Systematic molecular analyses of SHOX in Japanese patients with idiopathic short stature and Leri–Weill dyschondrosteosis. J. Hum. Genet. 2016, 61, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Verdin, H.; Fernández-Miñán, A.; Benito-Sanz, S.; Janssens, S.; Callewaert, B.; De Waele, K.; De Schepper, J.; François, I.; Menten, B.; Heath, K.E.; et al. Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci. Rep. 2015, 5, 17667. [Google Scholar] [CrossRef]
- Chen, J.; Wildhardt, G.; Zhong, Z.; Roth, R.; Weiss, B.; Steinberger, D.; Decker, J.; Blum, W.F.; Rappold, G. Enhancer deletions of the SHOX gene as a frequent cause of short stature: The essential role of a 250 kb downstream regulatory domain. J. Med. Genet. 2009, 46, 834–839. [Google Scholar] [CrossRef]
- Clement-Jones, M.; Schiller, S.; Rao, E.; Blaschke, R.J.; Zuniga, A.; Zeller, R.; Robson, S.C.; Binder, G.; Glass, I.; Strachan, T.; et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum. Mol. Genet. 2000, 9, 695–702. [Google Scholar] [CrossRef]
- Rappold, G.; Fukami, M.; Niesler, B.; Schiller, S.; Zumkeller, W.; Bettendorf, M.; Heinrich, U.; Vlachopapadoupoulou, E.; Reinehr, T.; Onigata, K.; et al. Deletions of the Homeobox Gene SHOX (Short Stature Homeobox) Are an Important Cause of Growth Failure in Children with Short Stature. J. Clin. Endocrinol. Metab. 2002, 87, 1402–1406. [Google Scholar] [CrossRef]
- Munns, C.J.F.; Haase, H.R.; Crowther, L.M.; Hayes, M.T.; Blaschke, R.; Rappold, G.; Glass, I.A.; Batch, J.A. Expression of SHOX in Human Fetal and Childhood Growth Plate. J. Clin. Endocrinol. Metab. 2004, 89, 4130–4135. [Google Scholar] [CrossRef][Green Version]
- Bunyan, D.J.; Baffico, M.; Capone, L.; Vannelli, S.; Iughetti, L.; Schmitt, S.; Taylor, E.J.; Herridge, A.A.; Shears, D.; Forabosco, A.; et al. Duplications upstream and downstream of SHOX identified as novel causes of Leri-Weill dyschondrosteosis or idiopathic short stature. Am. J. Med. Genet. Part A 2016, 170, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.A.L.; Souza, S.C.; Nishi, M.Y.; Billerbeck, A.E.; Libório, D.C.C.; Kim, C.A.; Arnhold, I.J.P.; Mendonca, B.B. SHOX mutations in idiopathic short stature and Leri-Weill dyschondrosteosis: Frequency and phenotypic variability. Clin. Endocrinol. 2006, 66, 130–135. [Google Scholar] [CrossRef]
- David, A.; Kun, Z.; Nyírő, G.; Szántó, Z.; Patócs, A. Heterozygous Deletion in Exons 4-5 of SHOX Gene in a Patient Diagnosed as Idiopathic Short Stature. CASE Rep. Acta Med. Marisiensis 2017, 63, 155–158. [Google Scholar] [CrossRef]
- Blaschke, R.J.; Rappold, G.A. SHOX in Short Stature Syndromes. Horm. Res. Paediatr. 2001, 55, 21–23. [Google Scholar] [CrossRef]
- Anton, J.I.; Reitz, G.B. Spiegel MB Madelung’s deformity. Ann. Surg. 1938, 108, 411–439. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, M.; Anton, J.I.; Spiegel, M.B. Madelung’s deformity: Consideration of its roentgenological diagnostic criteria. Am. J. Roentgenol. 1939, 42, 671–676. [Google Scholar]
- Dawe, C.; Wynne-Davies, R.; Fulford, G.E. Clinical variation in dyschondrosteosis. A report on 13 individuals in 8 families. J. Bone Joint Surg. Br. 1982, 64, 377–381. [Google Scholar] [CrossRef]
- Belin, V.; Cusin, V.; Viot, G.; Girlich, D.; Toutain, A.; Moncla, A.; Vekemans, M.; Merrer MLe Munnich, A.; Cormier-Daire, V. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat. Genet. 1998, 19, 67–69. [Google Scholar] [CrossRef]
- Shears, D.J.; Vassal, H.J.; Goodman, F.R.; Palmer, R.W.; Reardon, W.; Superti-Furga, A.; Scambler, P.J.; Winter, R.M. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat. Genet. 1998, 19, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Blaschke, R.J.; Marchini, A.; Niesler, B.; Burnett, M.; Rappold, G.A. The Leri-Weill and Turner syndrome homeobox gene SHOX encodes a cell-type specific transcriptional activator. Hum. Mol. Genet. 2001, 10, 3083–3091. [Google Scholar] [CrossRef]
- Grigelioniene, G.; Eklof, O.; Ivarsson, S.A.; Westphal, O.; Neumeyer, L.; Kedra, D.; Dumanski, J.P.; Hagenas, L. Mutations in short stature homeobox containing gene (SHOX) in dyschondrosteosis but not in hypochondroplasia. Hum. Genet. 2000, 107, 145–149. [Google Scholar] [CrossRef]
- Schiller, S.; Spranger, S.; Schechinger, B.; Fukami, M.; Merker, S.; Drop, S.L.S.; Tröger, J.; Knoblauch, H.; Kunze, J.; Seidel, L.; et al. Phenotypic variation and genetic heterogeneity in Leri-Weill syndrome. Eur. J. Hum. Genet. 2000, 8, 54–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Musebeck, J.; Mohnike, K.; Beye, P.; Tonnies, H.; Neitzel, H.; Schnabel, D.; Gruters, A.; Wieacker, P.F.; Stumm, M. Short stature homeobox-containing gene deletion screening by fluorescence in situ hybridisation in patients with short stature. Eur. J. Pediatr. 2001, 160, 561–565. [Google Scholar] [CrossRef]
- Thomas, G.P.L.; Johnson, D.; Byren, J.C.; Judge, A.D.; Jayamohan, J.; Magdum, S.A.; Richards, P.G.; Wall, S.A. Periodical shifts in the surgical correction of sagittal craniosynostosis. J. Neurosurg. Pediatr. 2015, 15, 348–349. [Google Scholar]
- Kaissi, A.A.; Chehida, F.; Ben Latos-Bielenska, A.; Gharbi, H.; Ghachem, M.; Ben Hendaoui, L.; Kozlowski, K. A novel form of ischio-vertebral syndrome. Skeletal. Radiol. 2007, 36, 77–81. [Google Scholar] [CrossRef]
- Cohen, P.A.; Kalifa, G.; Donoghue, V.; Adamsbaum, C.; Haddad, F.; Dubousset, J. Ischio-vertebral dysplasia: A distinct entity. Pediatr. Radiol. 1999, 29, 131–134. [Google Scholar] [CrossRef]
- Nishimura, G.; Kimizuka, M.; Shiro, R.; Nii, E.; Nishiyama, M.; Kawano, T.; Kaku, T.; Kawada, Y. Ischio-spinal dysostosis: A previously unrecognised combination of malformations. Pediatr. Radiol. 1999, 29, 212–217. [Google Scholar] [CrossRef]
- Nievergelt, K. Positiver Vaterschaftsnachweis auf grund erblicher Missbildungen der Extremitaten. Arch Klaus-Stift Vererb-Forsch 1944, 19, 157. [Google Scholar]
- Robinow, M.; Silverman, F.N.; Smith, H.D. A newly recognized dwarfing syndrome. Am. J. Dis. Child. 1969, 117, 645–651. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Kaissi, A.; Shboul, M.; Kenis, V.; Grill, F.; Ganger, R.; Kircher, S.G. Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan. Medicines 2019, 6, 60. https://doi.org/10.3390/medicines6020060
Al Kaissi A, Shboul M, Kenis V, Grill F, Ganger R, Kircher SG. Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan. Medicines. 2019; 6(2):60. https://doi.org/10.3390/medicines6020060
Chicago/Turabian StyleAl Kaissi, Ali, Mohammad Shboul, Vladimir Kenis, Franz Grill, Rudolf Ganger, and Susanne Gerit Kircher. 2019. "Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan" Medicines 6, no. 2: 60. https://doi.org/10.3390/medicines6020060
APA StyleAl Kaissi, A., Shboul, M., Kenis, V., Grill, F., Ganger, R., & Kircher, S. G. (2019). Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan. Medicines, 6(2), 60. https://doi.org/10.3390/medicines6020060